Pulmonary hypertension: a rare but important cause of dyspnoea
Breathing problems
Pulmonary hypertension can be a devastating disease that is easily missed in the early stages because of its typically nonspecific presentation with gradually increasing dyspnoea. With advances in management, most forms of pulmonary hypertension are treatable. Early diagnosis and treatment are key to improving functional and haemodynamic outcomes and survival.
Correction
A correction for this article will be published in the September 2019 issue of Respiratory Medicine Today. The online version and the full text PDF of this article (see link above) have been corrected.
- Pulmonary hypertension (PH) is characterised by an elevation in pulmonary artery pressure at rest and presents with progressive breathlessness as the cardinal symptom.
- PH can be associated with more than 50 different conditions, which are categorised into five groups based on pathophysiological mechanisms; accurate classification is crucial to management as treatment options and goals vary between groups.
- As symptoms of PH can be vague or nonspecific, diagnosis is often delayed, with patients typically progressing to advanced disease before diagnosis.
- Identifying patients early in the disease course can improve functional and haemodynamic outcomes.
- Group 1 PH (pulmonary arterial hypertension [PAH]) is rare; if it is suspected then prompt referral of the patient to a specialist with an expert interest in PH is recommended.
- Long-term management of patients with PAH, Group 4 (chronic thromboembolic) or Group 5 (unclear or multifactorial) PH should be delivered or co-ordinated through a designated PH clinic or service.
Pulmonary hypertension (PH) is a pathophysiological condition characterised by an increase in the mean pulmonary artery pressure. It is defined by a mean pulmonary artery pressure of 25mmHg or higher at rest, measured during right-heart catheterisation. A change to this threshold, to a mean pulmonary artery pressure of more than 20mmHg, was recently proposed.1 If untreated, PH can be a devastating condition with high morbidity, and mortality rates that are poorer than for many metastatic malignancies. Increased recognition of patients with PH, allowing early diagnosis and intervention, is the focus of current management campaigns to alter the natural history of this disease.
Classification of pulmonary hypertension
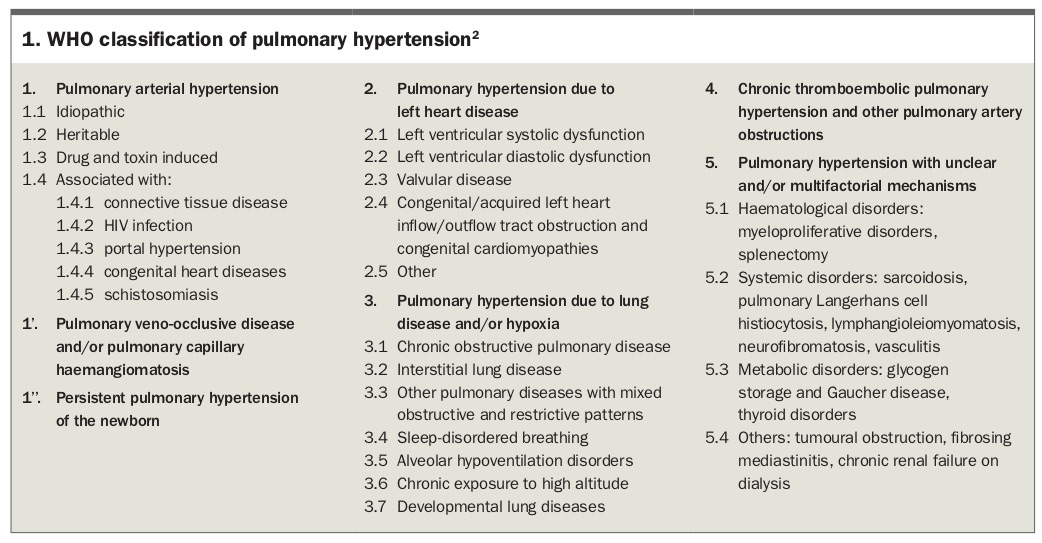
Conditions that cause PH are classified by the WHO into five major groups based on shared pathophysiological mechanisms, which guide the therapeutic approach. The WHO diagnostic groups are:
small pulmonary arteries leading to increased pulmonary vascular resistance are the primary abnormality
Conditions included in the five WHO diagnostic groups are listed in Box 1.2 These WHO diagnostic groups are distinct from the WHO functional class system (Classes I to IV), which was adapted from the NYHA functional class system.
{kind=link}
Accurate diagnosis of the PH group is crucial to ensuring management is targeted to the underlying pathophysiology. For example, selective pulmonary vasodilators are the only proven drug therapies for patients with PAH and Group 4 PH. However, their use to treat patients with PH in other groups may precipitate pulmonary oedema or other adverse cardiac events (Group 2 PH) or exacerbate hypoxia and breathlessness (Group 3 PH).
Group 1. Pulmonary arterial hypertension
PAH (Group 1 PH) is a progressive and often devastating condition. Proliferative changes in the lung microcirculation lead to increased pulmonary vascular resistance. If untreated, this results in right heart remodelling and ultimately right heart failure and death. Historically, patients with PAH had a grim prognosis, but in the past 15 to 20 years their functional status and life expectancy have improved significantly with major developments in targeted drug treatments.
Group 2. PH secondary to left heart disease
PH secondary to left heart disease (e.g. ventricular or valvular disease) is the most common type of PH. Treatment of the underlying disease and risk factor modification are key to management. Stiffness of the left ventricle (heart failure with preserved ejection fraction) is increasingly identified as a cause of PH.
Group 3. PH secondary to lung disease or hypoxia
PH secondary to lung disease, sleep-disordered breathing or hypoxia is relatively common. Optimising the underlying condition with appropriate therapy, supplemental oxygen and respiratory support is the key to management. Typically, patients do not benefit from targeted drug treatment.
Group 4. Chronic thromboembolic PH
Chronic thromboembolic PH is thought to occur secondary to vascular remodelling of the pulmonary arteries in the setting of mechanical obstruction by unresolved thrombus. The underlying pathophysiology is similar to that of PAH. Where possible, surgical intervention with pulmonary endarterectomy is the preferred treatment, as it has the potential to ‘cure’ PH. However, interventional radiological approaches or targeted drug therapy can be used in patients deemed unsuitable for surgery because of their pulmonary vasculature status, comorbidities or functional state.
Group 5. PH with unclear or multifactorial mechanisms
Group 5 PH includes conditions whose pathophysiology does not fit within Groups 1 to 4 PH. The mechanism driving abnormal pulmonary artery pressures in patients with Group 5 PH is often unclear. The most common cause of PH in this group is sarcoidosis.
Presentation
Clinical features of PH are nonspecific and include dyspnoea, fatigue, generalised weakness and, in later stages, chest pain, syncope and evidence of right heart failure. Given the nonspecific nature of these symptoms and the broad differential diagnosis, the diagnosis of PH is often overlooked, contributing to delayed management. Particularly in patients with PAH or Group 4 PH, opportunities are often missed to diagnose and treat disease at an early, potentially reversible stage before significant pulmonary vascular remodelling. A high index of suspicion is required as identifying patients early in the disease course and referring them to specialists with specific expertise in PH can improve functional and haemodynamic outcomes (see the case history in Box 2).3,4
{kind=link}
Diagnosis
Diagnosis of PH requires a comprehensive set of investigations to confirm that haemodynamic criteria are met and to identify the aetiology and severity of the condition. Involvement of a multidisciplinary team with expertise in cardiology, respiratory medicine, imaging and connective tissue diseases is recommended. Given the complexity of managing patients with PH, referral to a clinic with expert medical, nursing, pharmacological and psychological support is also recommended.
A diagnostic algorithm that highlights the complexities involved in accurate diagnosis of PH and the need for early referral and multidisciplinary team involvement is shown in the Flowchart.5
Transthoracic echocardiography
Patients with suspected PH are recommended to undergo transthoracic echocardiography. If tricuspid regurgitation is present then the systolic pulmonary artery pressure can be estimated. Isolated or severe dilation of the right ventricle or atrium may be a clue to PH if systolic pulmonary artery pressure cannot be estimated. GPs can help the supervising cardiologist by stating in the referral that PH is suspected. If possible, patients should be referred to an echocardiography service with experience investigating suspected PH. Stress echocardiography may provide additional information. However, occasionally PH is present and not detected by echocardiography.
• patients with systemic sclerosis
• first-degree relatives of patients diagnosed with hereditary PAH
• patients with portal hypertension who are candidates for liver transplantation.
Right-heart catheterisation
Right-heart catheterisation is needed for a definitive diagnosis of PAH and clarification of the prognosis, as well as to quantify pulmonary artery pressure. Accurate measurement of cardiac output and pulmonary capillary wedge pressure are essential in evaluation of left heart causes of elevated pulmonary artery pressure. In addition, vasoreactivity challenge testing with a short-acting pulmonary vasodilator (e.g. inhaled nitric oxide) can identify a small subset of patients with PAH who have significant vasoreactivity and are best managed with high-dose calcium channel blockers.
Management
Targeted drug treatments are available for patients with PAH and some cases of chronic thromboembolic PH. The management of patients with other types of PH is discussed above (see specific PH groups).
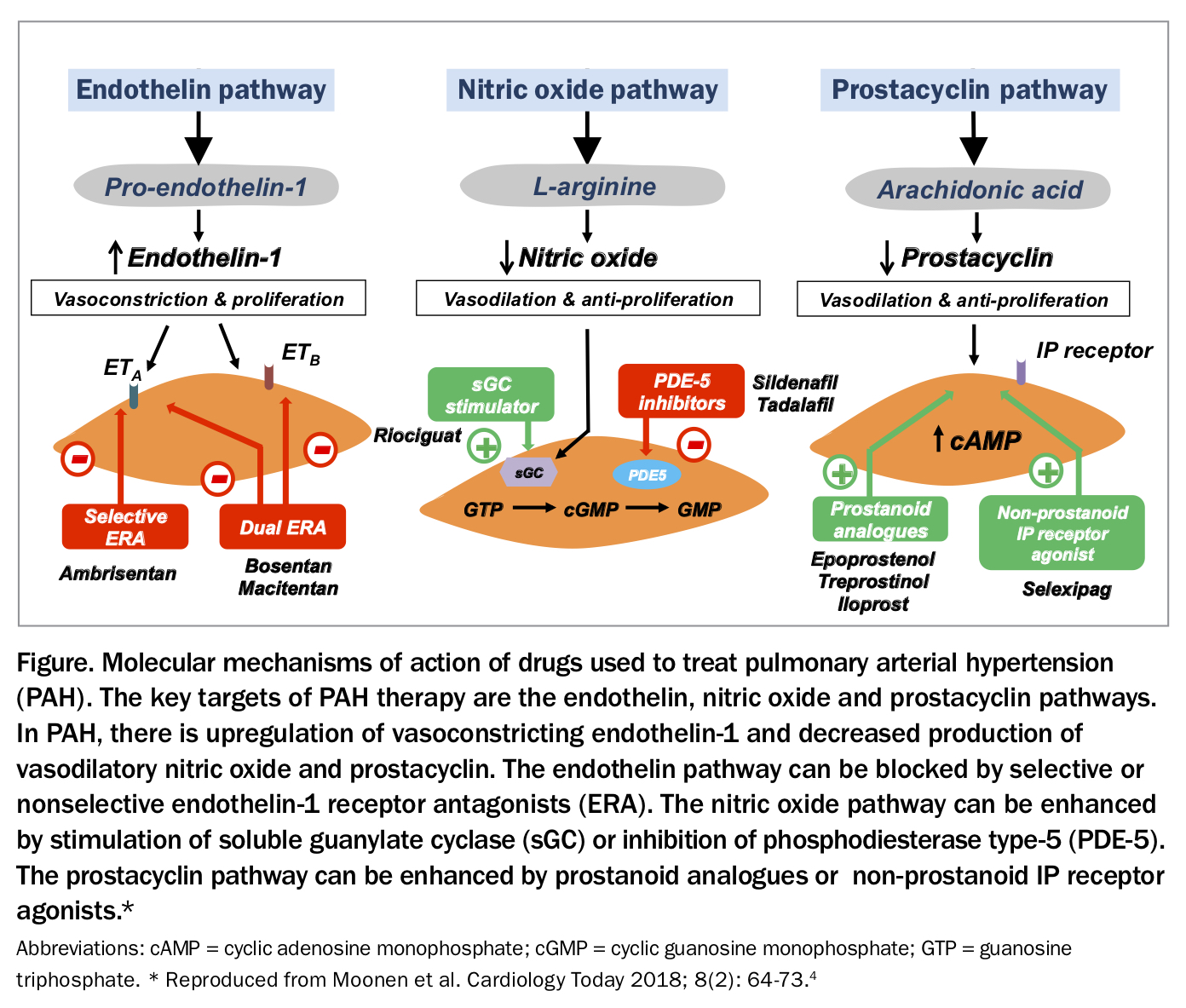
The targeted treatments comprise pulmonary vasodilators that target the endothelin, nitric oxide or prostacyclin pathways, which are involved in pulmonary vasoconstriction and dilation (Figure).5 These treatments fall into four major classes:
• phosphodiesterase type 5 inhibitors (e.g. sildenafil and tadalafil) – PAH
• soluble guanylate cyclase stimulators (e.g. riociguat) – PAH and Group 4 PH
• endothelin receptor antagonists (e.g. bosentan, macitentan and ambrisentan) – PAH
• prostacyclins and prostacyclin analogues (e.g. epoprostenol, iloprost and selexipag [the last is not PBS listed]) – PAH.
{kind=link}
In addition, calcium channel blockers may be useful in the small proportion of patients with PAH who show an acute vasodilator response on vasoreactivity challenge testing.
Most patients with PAH are managed with combination therapy, particularly if they have moderate or severe disease. Combination therapy is ideally instituted upfront or in a rapid stepwise fashion. The complexities of these medications and their interactions and side effects mean that patients are best managed through designated PH prescribing centres.
When and where to refer patients with PH
PH is rare and may coexist with more common causes of breathlessness (see the case history in Box 3). If there is evidence supporting the presence of a common cause of breathlessness then it is reasonable to treat this. A patient’s failure to respond rapidly as expected to treatment should raise a red flag for the clinician, indicating a need for referral. If PH is suspected then prompt referral to a specialist with an interest in PH is crucial (see the case history in Box 4).
{kind=link}
{kind=link}
Local expertise often determines the referral pathway. Both cardiologists and respiratory physicians, along with some rheumatologists, have expertise in the assessment and management of patients with PH. A small number of designated centres are approved to prescribe PH medications. Expedited referral to one of these centres should facilitate final investigation, diagnosis and treatment with appropriate medications. This maximises the therapeutic benefits and can provide informed supportive care to patients and their carers.
Conclusion
With advances in management, PH has evolved into a treatable disease with improved survival. Early diagnosis and treatment are key to improving functional and haemodynamic outcomes. However, delayed diagnosis of PH remains common. Diagnosis and management of patients with PH is complex and best undertaken in designated PH clinics. RMT