Not another exacerbation of COPD

COPD (chronic obstructive pulmonary disease)
Breathing problems
A 68-year-old recently retired male hairstylist with presumed chronic obstructive pulmonary disease presents with increasing dyspnoea and a persistent dry cough. Do his symptoms represent an exacerbation of COPD or some other condition?
- Spirometry is required for a diagnosis of chronic obstructive pulmonary disease.
- Diagnosis of interstitial lung diseases, including idiopathic pulmonary fibrosis (IPF), requires a multidisciplinary approach.
- IPF is an irreversible and progressive form of interstitial lung disease, with no known cause, that is associated with a poor prognosis.
- Antifibrotic therapies (pirfenidone and nintedanib) significantly slow disease progression by about 50% in patients with mild-to-moderate IPF.
- Early diagnosis of IPF is important so that antifibrotic therapy can be offered early to best preserve lung function.
- Supportive care is important in the management of patients with IPF and should be considered early, irrespective of antifibrotic therapy.
Case scenario
Fritz is a 68-year-old recently retired hairstylist who presents with increasing dyspnoea and persistent cough. He previously smoked about 30 cigarettes a day, for 44 years (i.e. 66 pack-years), but quit tobacco smoking two years ago when he was diagnosed clinically with chronic obstructive pulmonary disease (COPD). The diagnosis was not confirmed on spirometry. His symptom at the time was cough, and he started taking inhaled tiotropium bromide 18 mcg daily. He has also been taking lisinopril 20 mg daily for hypertension for the past five years. Fritz is overweight, with a body mass index of 30 kg/m2. He has been investigated for coronary artery disease as a contributor to his breathlessness; angiography showed 30% blockage in the mid left anterior descending artery and some minor small vessel disease.
Fritz experienced a presumed infective exacerbation of COPD nine months ago and, although his condition improved, he continued to have a persistent dry cough. On this occasion, he presents to you with a six-month history of increasing exertional dyspnoea and says he has had to give up playing golf. His respiratory rate at rest is 18 breaths per minute and oxygen saturation on room air is 94%. He appears comfortable at rest and there is no use of accessory muscles of respiration. On examination, there are some basal crackles that do not clear with coughing. His jugular venous pressure is not elevated and there is no peripheral oedema.
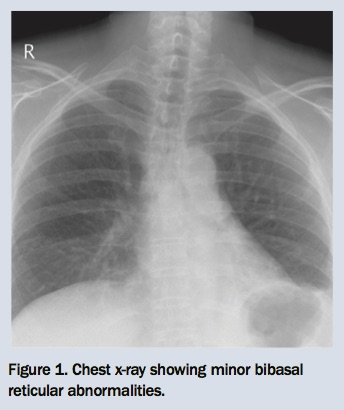
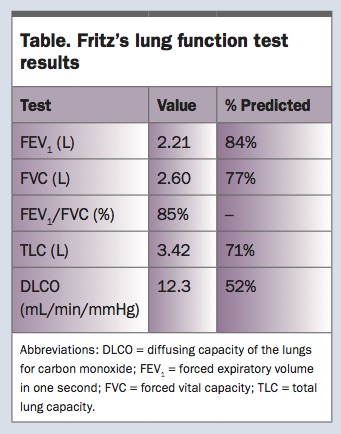
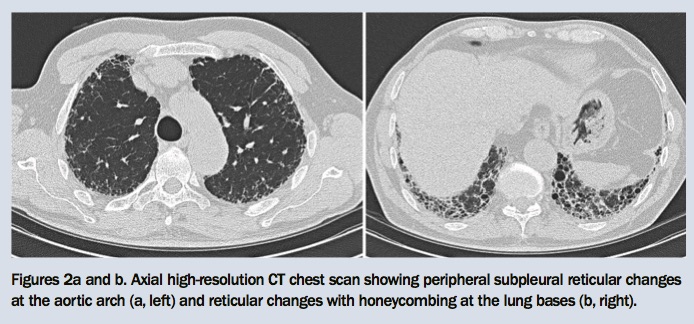
You refer Fritz for a chest x-ray and lung function tests. The chest x-ray shows minor bibasal reticular changes and reduced lung volumes (Figure 1). Results of the lung function tests show a mild restrictive ventilatory defect and moderately reduced gas transfer measurement (Table). After reviewing these results, you organise a high-resolution CT scan of the chest, which shows predominantly bibasal subpleural reticular changes with honeycombing (Figures 2a and b).
{kind=link}
{kind=link}
{kind=link}
What condition does Fritz have? Was his COPD diagnosis correct? When should an alternative pulmonary condition be suspected in primary care and how should it be managed?
Commentary
Fritz’s combination of symptoms, signs and lung function test results is consistent with an interstitial lung disease process. The lack of any clear cause, together with typical high-resolution CT chest scan findings, further narrows this down to a diagnosis of idiopathic pulmonary fibrosis (IPF). IPF is a chronic form of interstitial lung disease (ILD) characterised by irreversible and progressive fibrosis. As its name implies, the cause is not known, but several possible risk factors, including inhalation of irritants (e.g. cigarette smoke, wood and metal dusts and hair sprays), have been described. Although relatively rare, its incidence is increasing. IPF typically affects people over the age of 60 years and is slightly more common in men than women.
Prognosis is poor, with an average survival of three years from the time of diagnosis. This is likely to improve with the advent of two antifibrotic drug therapies, pirfenidone and nintedanib, that are now available on the PBS in Australia. Early and accurate diagnosis is key to optimising the outcome for affected individuals.
Diagnosis
For various reasons, many patients with IPF are diagnosed late in their disease, with an average lag of two years from the time of presentation. Gradually increasing exertional dyspnoea and a dry cough over months to years are typical presenting complaints. Patients often describe a chest infection as a starting point, but it is likely that the IPF was already present and the chest infection has merely unmasked the underlying condition. Many patients present late, having attributed their symptoms to getting old or being unfit, while doctors frequently ascribe the nonspecific symptoms to other, more common, medical conditions such as asthma, COPD or congestive heart failure. Comorbidities, including COPD and ischaemic heart disease, are also common in patients with IPF, which can increase the delay to diagnosis.
It is important to take a careful history to rule out known causes of pulmonary fibrosis. The presence of bibasal crackles on chest auscultation is an important early sign of IPF, and digital clubbing is present in up to half of patients with IPF. Chest x-ray changes, when present, are nonspecific and are often subtle in the early stages of disease. A restrictive ventilatory defect and reduced gas transfer measurement are seen on lung function testing. However, in early disease, an isolated reduction in gas transfer might be the only physiological abnormality. Findings from bedside spirometry should be confirmed and further investigated with full lung function tests, including lung volume and gas transfer measurements.
The presence of crackles on chest auscultation and an abnormal chest x-ray raise the possibility of pulmonary fibrosis, but a high-resolution CT scan of the chest is necessary to secure the diagnosis. Bibasal subpleural reticular changes and honeycombing are typical abnormalities of IPF seen on CT. Ground glass changes are typically absent.
Patients suspected of having IPF should be referred to a respiratory physician for confirmation of the diagnosis and management. An accurate diagnosis is necessary to inform prognosis and treatment options. It is equally important to correctly diagnose other forms of ILD (Flowchart) because the prognosis for these conditions is invariably better and the treatment options, such as avoidance of triggers and consideration of immunosuppressive therapies, are distinctly different from those for IPF. As ILDs share many similar clinical, radiological and histological features, a multidisciplinary team approach, considering all these findings, is now regarded as the gold standard for diagnosis of ILD.1
Treatment
The antifibrotic medications pirfenidone and nintedanib have both been shown to slow disease progression by about 50% in patients with mild-to-moderate IPF (patients with severe disease were not studied).2,3 Compared with patients with moderate disease, those with mild disease had the same rate of disease progression and the same response to treatment.4,5 Although IPF is a fatal disease, the disease course is variable, with some patients experiencing rapid deterioration and others having a slower decline. Some patients may also demonstrate a step-like clinical course, with periods of stability alternating with periods of deterioration.
As there is no sure way of predicting how any individual’s disease will behave, offering drug treatment as early as possible affords patients the best chance of preserved lung function. Patients must meet strict PBS criteria to access pirfenidone or nintedanib, which can only be prescribed by a respiratory or other specialist physician. The choice of which drug to use is discussed with patients and is largely based on side effect profile. Gastrointestinal side effects, including nausea, diarrhoea and loss of appetite, are common. Liver toxicity, although rare, should be actively investigated with routine liver function tests. Diarrhoea is more common with nintedanib, while a photosensitive rash is more common with pirfenidone. Side effects can be managed with dose reductions and, in some cases, a temporary break from the drug.
Supportive care
Despite these therapeutic advances, the disease course will continue to progress during treatment and patients with IPF often do not feel better. Therefore, it is imperative that supportive measures are instituted in parallel. These include treatment of symptoms with palliative care, addressing the common comorbidities of anxiety and depression, ensuring the patient is up to date with vaccinations, has a good diet and maintains physical activity, and making advance care plans when the patient is well enough to make informed choices.
Symptomatic and palliative care can be initiated by the treating doctor, or the patient can be referred to a palliative care service. Intractable breathlessness, an inevitable symptom of IPF, is particularly devastating. Although there is no evidence that any symptomatic treatment works in patients with ILD, various measures including using a hand-held fan, supplemental oxygen and morphine can be tried. Exercise training has been associated with improved walking distance and health-related quality of life, regardless of disease severity, in patients with ILD.6 It is therefore important to consider pulmonary rehabilitation for all patients where possible.
Patients might also benefit from support groups that can be accessed through the Lung Foundation Australia.7 A more comprehensive review of IPF management is given in the latest guideline statement from the Thoracic Society of Australia and New Zealand and the Lung Foundation Australia.8 Lung transplantation may be a viable option for a select few patients.
Conclusions
IPF remains a lethal disease and there is much ongoing research aimed at improving therapeutic options. Unfortunately, many patients are diagnosed late in their disease course. It is crucial that IPF is recognised and patients are referred early to achieve the best possible outcome. Antifibrotic medications are now available on the PBS. Although drug side effects are common, they are often manageable. However, drug cessation should be considered if the side effects significantly affect quality of life. Discussions about stopping antifibrotic therapy should be had in conjunction with specialists. Supportive measures, in addition to drug treatment, are important in the management of IPF. RMT