Paediatric cystic fibrosis. Part 1: diagnosis

Cystic fibrosis
Lung diseases
Pathways to diagnosing cystic fibrosis can occur at different stages. Some children will have gene mutations not detected by newborn screening in Australia, and GPs need to remain alert to clinical presentations of the disease. GPs are also in a prime position to provide genetic counselling for carriers and to promote carrier screening in the community.
- Despite the newborn screening program in Australia, rare genetic mutations may be missed and clinical presentations of cystic fibrosis (CF) can occur.
- Patients presenting at any age can have milder forms of CF or CF-related disease.
- Carrier screening for CF is readily available and can be offered to couples planning pregnancy, even if there is no family history of CF.
- Carriers and their families need genetic counselling, which GPs can provide.
Cystic fibrosis (CF) is the most common life-shortening genetic disorder in children. Its live birth prevalence is one in 3000, with more than 3400 people living with CF in Australia.1 Over recent decades, life expectancy and quality of life for people living with CF have improved as the result of early diagnosis and subsequent intensive management. Although the median life expectancy remains at only 37 years, the estimate for a baby born with CF today is 50 years.2 However, this estimate was calculated before the development of new drugs that activate the CF transmembrane conductance regulator (CFTR), which are expected to substantially influence outcomes, including maintaining lung function and nutrition and reducing sweat chloride levels, for people with CF. Although CF care is provided by a multidisciplinary specialist team, primary care physicians are an integral part of the cohesive care delivered.
This article – the first in a series covering issues in CF care, with an emphasis on children – focuses on diagnosis. Pathways to diagnosing CF can occur at different stages throughout the lifespan. Subsequent articles will cover respiratory care, nutrition and gastroenterological complications and other matters (e.g. CF-related diabetes, musculoskeletal complications, fertility, adolescence and transition to adult care).
Aetiology
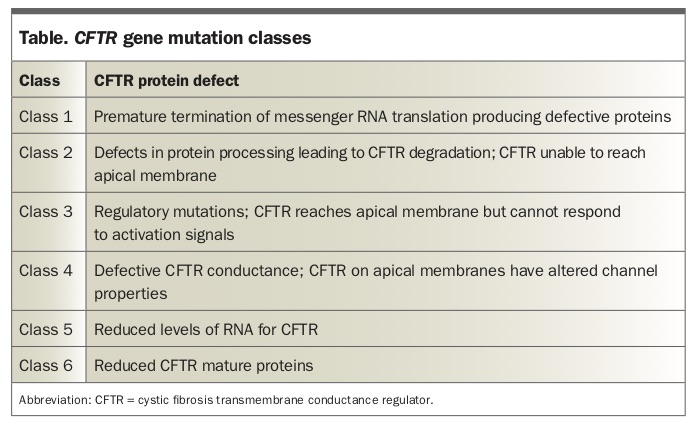
CF is an autosomal recessive disorder caused by gene mutations on chromosome 7 encoding for the CFTR, a chloride channel that is expressed on the apical surface of epithelial cells in the airways, pancreatic ducts, gastrointestinal tract, bile ducts, vas deferens and sweat ducts. There are more than 2000 mutations that have been classified into six classes of CFTR protein defects (Table), a model that is helpful when thinking about the range of severities of clinical presentation and the action of new therapeutic agents.
{kind=link}
In general, class 1 to 3 mutations are associated with no functional CFTR protein and ‘classic’ CF, with suppurative lung disease, pancreatic exocrine insufficiency and elevated sweat electrolyte levels. Class 4 to 6 mutations are more variable but are often associated with residual CFTR function and a less severe phenotype (e.g. pancreatic-sufficient CF). Lung genotype to phenotype associations are weak.3 Some patients may not be classified as having CF, particularly in the absence of any lung-related complications, and will instead be deemed to have a CFTR-associated disorder.
Antenatal and carrier screening
An estimated one in 25 people of Caucasian or Ashkenazi Jewish descent carry a pathogenic CFTR gene mutation.4 Carriers are healthy, and most babies born with CF have no family history of the disease, which is typical for recessive conditions. Carrier screening for the most common CFTR gene mutations in Australia can be done using saliva or blood samples, and the results of this screening give families reproductive options. GPs are well placed to provide carrier screening, ideally before pregnancy to offer prospective parents the most options or in the early stages of pregnancy. Referral to a genetic counsellor can be considered when CFTR gene mutations are detected.
CFTR screening panels usually include a selection of the most common mutations, with a sensitivity of about 90%. Most of the more than 2000 CFTR gene mutations are very rare, but there is a small residual risk that a person who tests negative is a carrier of a mutation not included in the screening test. A further benefit of screening is that some screening programs offer tests for additional diseases, such as spinal muscular atrophy and fragile X syndrome, and data from these programs suggest that one in 20 women will be a carrier of one of these conditions.5
If prospective parents are identified as carriers before becoming pregnant, the option of in vitro fertilisation with preimplantation genetic diagnosis can be offered. If identified as carriers during pregnancy, antenatal diagnosis can be performed with chorionic villus sampling at 10 to 12 weeks’ gestation or by amniocentesis at 14 to 16 weeks. Some fetal antenatal scans show an echogenic bowel, which can be a feature of CF.
Newborn screening
Most babies with CF in Australia are diagnosed by newborn screening using blood spots collected on a Guthrie card two to four days after birth (Flowchart). The initial screening test measures immunoreactive trypsinogen level, and babies with levels above the 99th percentile of results will then have CFTR gene mutation analysis from the original blood spot.6 The mutations included in the screening test differ between states and territories.
Infants with two mutations are diagnosed with CF and are referred to a CF centre. Infants with one identified CFTR mutation may either be healthy carriers or have a second unidentified mutation and will require a sweat chloride test to determine which. Those with a positive result from this test will be referred to a CF centre, whereas the parents of those identified as carriers should receive genetic counselling.
Infants without any identified mutation are not tested further. However, some of these infants may have CF from two mutations that were not included in the screening test and will present later with clinical features. About 5 to 10% of infants with CF are missed by newborn screening, and GPs should remain suspicious about the diagnosis when children present with clinical features of the disease.
Childhood clinical presentation
CF in neonates most often presents as meconium ileus (in 20% of babies with CF), with infants presenting soon after birth with bowel obstruction or delayed (more than 48 hours) passage of meconium. Prolonged jaundice is also a possible presentation in neonates. Infants may present later with failure to thrive and steatorrhoea (pancreatic exocrine insufficiency) or recurrent lower respiratory infections (productive sounding cough or wheeze). Children rarely present with rectal prolapse or nasal polyps.
The best diagnostic investigation is a sweat chloride test. This is a specialised investigation that should be performed at an accredited CF care centre (usually located at tertiary children’s hospitals) to ensure accurate results. Genetic testing, although more widely available, may miss CF cases involving uncommon mutations. GPs can contact clinicians who specialise in CF for assistance with this diagnostic process.
Late clinical presentation
Occasionally, patients may present later in childhood or in adulthood. Sometimes these patients have pancreatic exocrine insufficiency (usually presenting in the first 12 months of life), but more often they are pancreatic sufficient and have CFTR gene mutations that maintain some function (i.e. chloride transport).
Recurrent lower respiratory tract infection is the most common late presentation. Some patients have CFTR-related disorders, such as congenital bilateral absence of the vas deferens, recurrent sinusitis or recurrent pancreatitis, rather than CF. A summary of clinical manifestations consistent with CF is shown in Box 1.7 In the absence of pulmonary or pancreatic exocrine dysfunction, patients would not usually qualify for a diagnosis of CF and would instead be classified as having a CFTR-related disorder. However, it is best to arrange a sweat chloride test when there is a suspicion of CF, as CFTR genotype analysis may miss patients with uncommon mutations.
{kind=link}
Conclusion
GPs should be aware of the newborn screening process and remain alert for individuals with CF missed by routine screening. CF carrier screening can be offered to women or couples who are planning a pregnancy or in the early phases of pregnancy. GPs may feel comfortable providing genetic counselling to carriers themselves or can refer them to a genetic counsellor if not. After receiving a diagnosis of CF, families require significant holistic support, and the familiar face of a family practitioner can be a great asset in helping them feel comfortable and supported. This support will be needed lifelong and will help the family to integrate the challenges of CF into the usual challenges of children growing up. Some useful CF resources for GPs are listed in Box 2.
{kind=link}
The next article in this series will cover the pulmonary management of CF and the new medications that activate CFTR. RMT