Paediatric cystic fibrosis. Part 3: Nutrition and gastrointestinal health
Dr Pham is a Paediatric Respiratory Fellow, Professor Massie is Paediatric Respiratory Consultant and Ms Harris is Paediatric Dietitian at the Royal Children’s Hospital, Melbourne, Vic.
Cystic fibrosis
Lung diseases
Management of nutrition, growth and gastrointestinal symptoms remain an integral part of care in patients with cystic fibrosis. The main nutritional issues are pancreatic exocrine insufficiency with secondary malabsorption, salt depletion, micronutrient deficiency and elevated metabolic requirements. Abdominal pain is also a common presentation and the cause can be difficult to diagnose.
- Pancreatic insufficiency leads to macronutrient maldigestion and impaired weight gain in children with cystic fibrosis (CF), particularly if enzymes are inadequately replaced.
- Patients with CF have an increased recommended daily caloric intake compared with healthy peers.
- The goal for CF nutritional care is to maintain a normal growth pattern.
- Excessive salt loss can lead to hyponatraemic, hypochloraemic dehydration with young children with CF at higher risk. Daily salt replacement is required.
- Gastrointestinal manifestations are common. There are some particular CF-specific diagnoses of abdominal pain to add to the usual differential diagnoses considered for a child without CF.
- Almost all severe cases of CF liver disease are detected within the first two decades of life.
Cystic fibrosis (CF) implicates almost every organ system, either directly or because of secondary consequences of the disease. This includes the gastrointestinal system and nutritional status. The cystic fibrosis transmembrane conductance regulator (CFTR) protein is expressed in the pancreatic ducts, intestinal mucosa and bile ducts. There is a baseline increase in metabolic rate causing a person with CF to require additional nutritional intake above the recommended daily allowance. The metabolic burden of suppurative lung disease adds to this further. Although we now recognise the disease burden of CF on the lungs, it was historically recognised as a lethal cause of malabsorption and originally called ‘fibro-cystic disease of the pancreas’. Close monitoring and management of nutrition, growth and gastrointestinal symptoms remain an integral part of CF care. There are positive correlations between nutritional status and lung health.
This article reviews the nutritional, gastro-intestinal and hepatic issues faced by patients with CF. The common manifestations, diagnostic algorithms and approaches to management are discussed. For all issues described, GPs are encouraged to work in liaison with the CF multidisciplinary team for continuity of care. Concerns can be addressed via phone discussions or joint telehealth reviews to allow for prompt initiation of management plans and appropriate decisions regarding disposition of care.
Nutrition
The main issues with nutrition for patients with CF are pancreatic exocrine insufficiency with secondary malabsorption, salt depletion, micronutrient deficiency and elevated metabolic requirements.
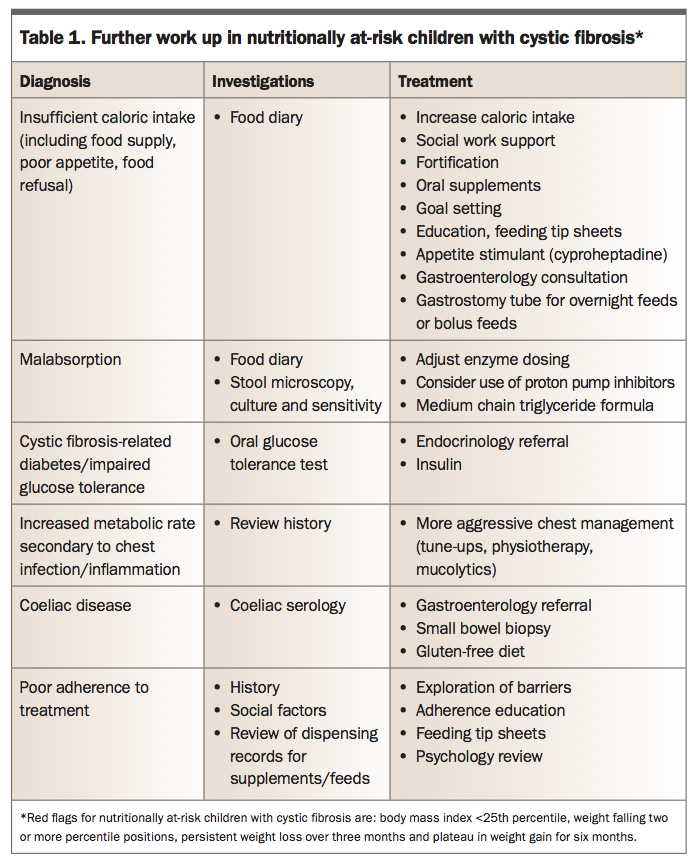
Better nutrition, as measured by body mass index (BMI), correlates positively with lung function (measured by forced expiratory volume in one second, FEV1) and negatively with rates of respiratory exacerbations. As such, care is delivered by a multidisciplinary team, including dietitians and gastroenterologists who regularly optimise the nutritional status of people with CF. Regular contact is maintained through three-monthly clinic visits and an annual nutritional assessment, with investigations including blood tests for measurement of fat soluble vitamins (A, D, E and K), iron and zinc levels. Nutritionally at-risk patients require further work up (Table 1).
{kind=link}
Pancreatic exocrine insufficiency
Pancreatic exocrine insufficiency affects about 85% of people with CF.1 Pancreatic status is closely related to the underlying CFTR gene mutation. Patients with two mutations from classes 1 to 3 have no functional CFTR at the epithelial surface of the pancreatic ducts and invariably have pancreatic exocrine insufficiency. It is thought that absent CFTR channel function impairs chloride and bicarbonate transport into the pancreatic ducts, leading to inspissated acidic pancreatic secretions. This altered environment leads to precipitation of the secreted pancreatic proteins, such as trypsinogen, causing pancreatic duct obstruction and secondary damage to the pancreatic acini. This process begins in utero with most (60%) infants exhibiting pancreatic exocrine insufficiency at birth, with the majority becoming pancreatic insufficient during the first year of life. There is overflow of pancreatic enzymes such as trypsinogen into the blood, which is the basis for the newborn screening test measuring trypsinogen level by immunoreactive assay (see Part 1 in this series). There is a progressive (painless) inflammatory process causing cumulative destruction of pancreatic acini that results in both exocrine (acinar) and eventually endocrine (islet cell) components being affected. CF-related diabetes will be discussed in the following article (Part 4) of this series.
About 15% of people with CF remain pancreatic sufficient, although their pancreatic function is still reduced compared with nonCF peers.1 However, this proportion may well increase with newer medications that improve CFTR function (correctors). These patients usually have CFTR gene mutations class 4 to 6 (see Part 1 in this series), which are associated with residual CFTR function. Since only 1 to 2% of residual pancreatic exocrine function is needed to maintain digestion of carbohydrate, protein and fat, these patients do not need pancreatic enzyme replacement therapy (PERT). About 20% of patients with CF who are pancreatic sufficient develop recurrent, painful pancreatitis. Of interest, many patients with idiopathic chronic pancreatitis are found to have one CFTR gene mutation. Presumably the 50% reduction in pancreatic CFTR function found in carriers, along with another risk factor (e.g. bile stones, alcohol use) is enough to cause pancreatitis.
In individuals with pancreatic exocrine insufficiency, the lack of pancreatic enzymes reaching the duodenum leads to mal-digestion of ingested fats, proteins and carbohydrates. Clinically, patients experience steatorrhoea (offensive, fatty, oily stools that do not flush away). This macronutrient depletion leads to failure to thrive, which is often first recognised by babies not regaining birth weight by 1 to 2 weeks of age. Investigations for pancreatic insufficiency include faecal microscopy for fat globules (undigested fat) but the gold standard is measurement of faecal pancreatic elastase levels (a pancreatic glycoprotein that is reduced in the stools of CF patients with pancreatic exocrine insufficiency and is a surrogate marker for other pancreatic enzyme levels).2
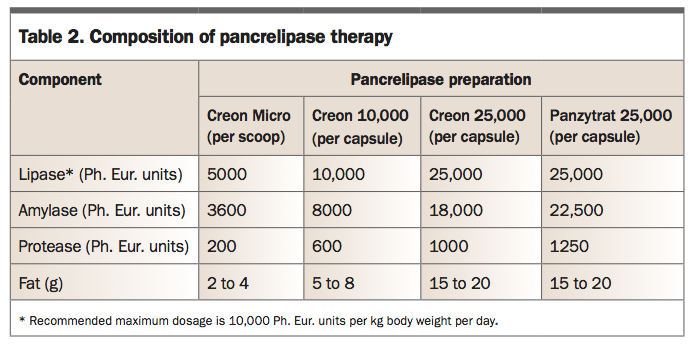
The management of patients with CF who have pancreatic exocrine insufficiency is with PERT with all meals and snacks. This is available as pancrelipase, a porcine pancreatic extract encapsulated in enteric-coated microspheres, in preparations appropriate for the developmental age and biological requirements of the child (Table 2). Infants can have their microspheres delivered in a spoonful of apple puree, although most will not start solids until 4 to 6 months of age. The microspheres are similar in size to food particles, mix homogeneously with chyme and remain inactive in the acidic gastric environment. Once they pass into the alkaline pH of the duodenum, the enteric coating rapidly dissolves, releasing enzymes for activation. If children take more than half an hour to eat their food, enzymes should be taken again. Acid reducing agents such as protein pump inhibitors or H2-antagonists can be used to optimise duodenal PERT activation when PERT dosing exceeds recommendations.
{kind=link}
Poor adherence to PERT is the most common reason for breakthrough steatorrhoea or poor growth. Children are usually able to take their enzymes as enteric-coated capsules by school age, but reluctance to take them at school in front of friends and teachers is a common problem. GPs are well placed to work with children and families to enhance adherence.
Salt depletion
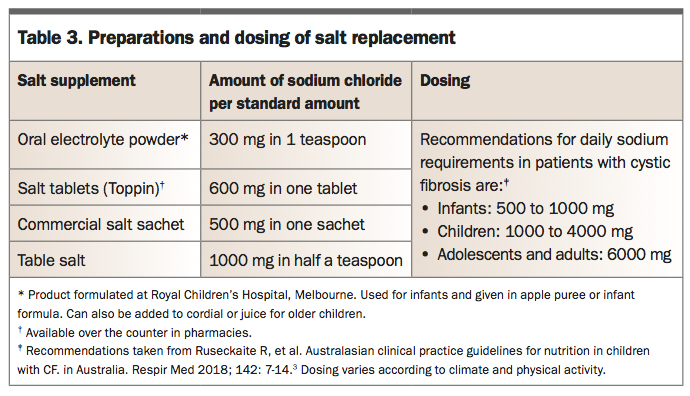
Individuals with CF lose excessive salt through their skin (see Part 1 of this series) and risk dehydration, particularly infants given their higher body surface area. Before newborn screening this was a serious and potentially fatal complication of CF. It can still occur, either in babies missed by screening or in older patients who do not take appropriate salt replacement. There is an unusual biochemical pattern of hyponatraemic, hypochloraemic metabolic alkalosis. The most common, mild presentation is muscle cramps. Low-grade salt depletion can also be a cause of growth failure. Daily replacement with salt supplementation is essential for all patients with CF (Table 3).3 It is recommended that most children with CF use liberal table salt on food (use iodised salt as iodine deficiency is common in many parts of Australia). Infants can have salt or oral rehydration solution added to their bottles or in apple puree with their PERT. Older children can use salt tablets or gel caps filled with table salt. Sports drinks alone do not provide sufficient additional salt for patients with CF.
{kind=link}
Micronutrient deficiency
Fat soluble vitamins (A, D, E and K), certain minerals (iron) and trace elements (zinc) are also poorly absorbed in patients with CF (Table 4). Now that most babies are diagnosed through newborn screening, it is rare to see patients with complications of vitamin deficiency. Iron deficiency in CF can be multi-factorial, secondary to poor absorption, increased iron loss in sputum or the gastrointestinal tract, and secondary to chronic disease.
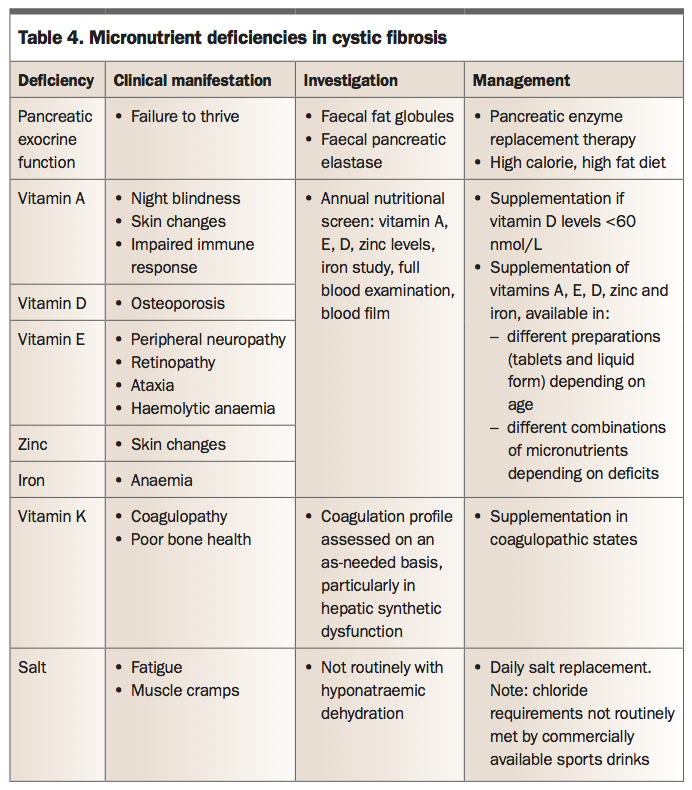{kind=link}
Metabolic requirements and nutritional advice
Children with CF have increased metabolic demands. This is partly a result of absent CFTR function, presumably because other epithelial transport proteins are working overtime, but there is no clear mechanism described. In addition to this are the metabolic requirements of chronic pulmonary infection, pulmonary exacerbations and other comorbidities, such as CF-associated liver disease and CF-related diabetes (discussed in the next part of this series). As a baseline, children with CF are encouraged to ingest 120% of the recommended daily intake of calories but once suppurative lung disease develops, this could be as high as 200%.4 Inflammatory cytokines (e.g. Il-1) induce appetite loss and there can be the added complexity of psychological issues impacting on eating behaviours. Despite this, the goal of CF nutritional care is to maintain a normal growth pattern, understanding that decreased early mortality has been associated with greater body weight and BMI in patients with CF.5 Regular monitoring of anthropometric measurements on growth charts helps identify at-risk patients warranting further review (Table 1). GPs are well placed to help children and families with these assessments by working in conjunction with the CF dietitians and medical teams, flagging concerns and supporting management plans.
Behavioural feeding problems are common in young children who have ‘picky’ eating patterns and whose parents are worried about nutrition. Behavioural strategies around eating and mealtimes work well in the younger age groups. These can include encouraging regular meals, praising good eating habits and establishing rules around mealtimes. A variety of oral nutritional supplements are available through the recommendations of CF dietitians. Appetite stimulants such as cyproheptadine allow for increased weight and appetite demonstrated by small short-term trials.6 Gastrostomy tubes are offered to children with sustained poor weight gain (BMI <10 to 25%) once other options are exhausted. This facilitates supplementary feeding, either as food boluses throughout the day or through overnight feeding regimens. At times of nutritional failure, short-term parenteral nutrition may be considered.
CFTR modulators have been documented to improve nutrition (see Part 2 of this series).7 This is thought to be through better gastrointestinal absorption and perhaps a lower metabolic rate. Given that most patients with CF have pancreatic exocrine insufficiency from birth or soon after, CFTR modulators do not improve pancreatic exocrine function. Perhaps, in the future, starting CFTR modulators from birth (or even in utero) could preserve pancreatic exocrine function. This would need carrier screening in the community (see Part 1 of this series) to identify affected fetuses or babies for treatment in the first few days of life.
Gastrointestinal tract
Gastrointestinal problems are common in patients with CF as the CFTR is expressed in the epithelial cells of the gut. Meconium ileus, distal intestinal obstruction syndrome, constipation and recurrent abdominal pain are the main features. These are all variants of the same pathology, that is, poor intestinal secretion of electrolytes. Other problems also occur with a greater frequency than expected: intussusception (thickened secretions adherent to bowel wall forming lead points can catch in intestinal peristalsis and cause the invagination of bowel segments), coeliac disease (no explanation), appendicitis (normal incidence, but contrary to early teachings that suggested it did not occur) and in older patients, gastrointestinal adenoma (potentially from chronic inflammation). These can all make the differential diagnosis of abdominal pain in a child with CF difficult. Potential aetiologies of abdominal pain in CF are summarised in the Box.8-12 An approach for navigating mild chronic abdominal pain is presented in the Flowchart.13 The pragmatic approach is that if there is no more obvious cause of abdominal pain, then pursue constipation or distal intestinal obstruction syndrome (DIOS) as the culprit and treat this. Ongoing pain, despite effective gut wash-out will be something else. When managing children with CF presenting with acute abdominal pain, all the same differential diagnoses should be considered as for a child without CF. It is also encouraged that management of gastro-intestinal complications are discussed with the CF team to help guide disposition and lines of management.
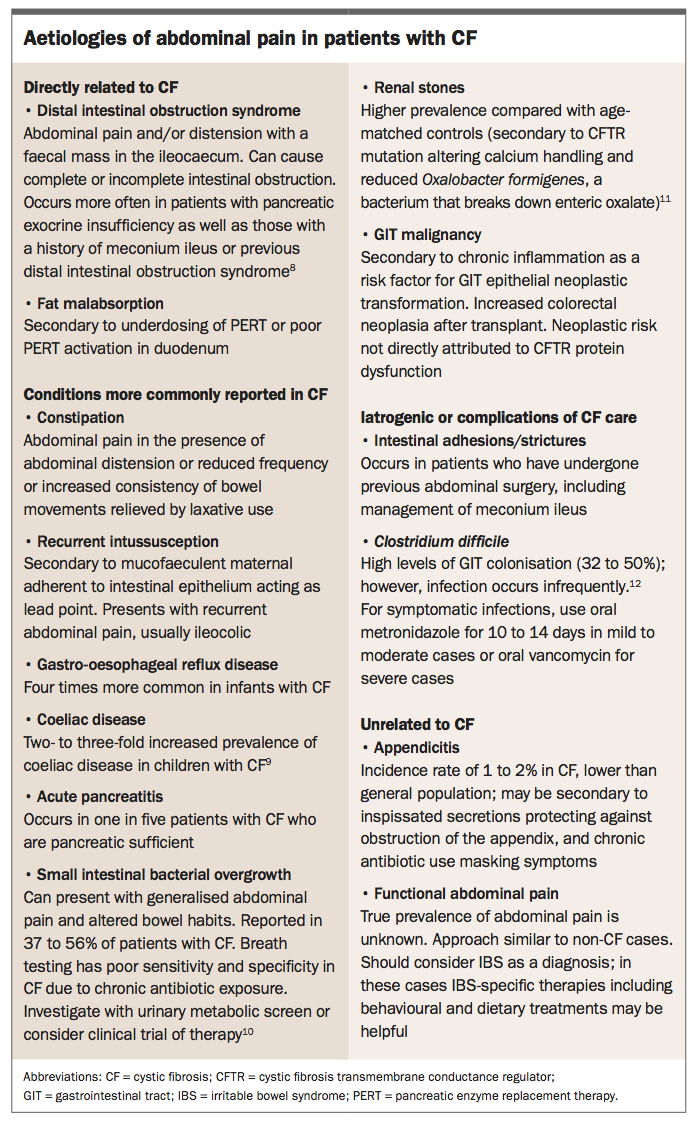{kind=link}
Meconium ileus
The earliest presentation of gastrointestinal manifestations is meconium ileus, which occurs in approximately 20% of babies with CF.14 This is a neonatal emergency and is predominantly recognised within 48 hours with intestinal obstruction (abdominal distension and vomiting) and failure to pass meconium. Babies with meconium ileus need urgent transfer to a neonatal intensive care unit with surgical facilities. About half will respond to medical treatment with gastrograffin wash-out, the other half requiring surgical removal of inspissated meconium. Some will require a temporary ileostomy. Gastrointestinal surgery increases the risk of abdominal adhesions which can cause bowel obstruction or be the lead point for intussusception. Occasionally the terminal ileum requires resection and this may have implications for nutrition and the development of intestinal function later on.
Distal intestinal obstruction syndrome and constipation
Altered CFTR function leads to the composition of intestinal fluid being more viscous, which may cause bowel obstruction (DIOS) due to faecal impaction of the colon or recurrent constipation. This is by far the most common cause of severe abdominal pain in children with CF. DIOS is most common in the early school years with a notable risk factor being poor adherence with PERT. Patients present with colicky, central abdominal pain, reduced (or no) passage of stool and sometimes vomiting. Constipation is a milder version of this and presents in a more insidious manner. It is occasionally complicated by overflow diarrhoea which can confuse the clinical picture. An abdominal x-ray may be useful in demonstrating bowel obstruction, but faecal loading is common in CF so an x-ray is not always conclusive.
Most patients with DIOS can be managed in the outpatient setting, under the guidance of the CF team. GPs can help facilitate this process by offering regular clinical reviews and liaising with the CF team. Rarely, a nasogastric tube is required for problematic vomiting. The key is ingestion of a good dose of aperient such as macrogol (suggested dosing of 2 g/kg daily, and titrated as per response to a maximum of 100 g daily), repeated regularly until there has been a passage of solid stool, followed by watery fluid (of macrogol and stool). If not responding within one week, then treatment is escalated. Rarely, more significant interventions such as enteral gastrograffin or enemas are required. Ideally, surgery is avoided because of the risk of adhesions developing that can lead to further bouts of DIOS. Patients and families who have experienced DIOS often recognise its subsequent onset and use macrogol early. Some find their way to regular doses of macrogol or other agents such as lactulose or paraffin (or a ‘cocktail’ of agents that result in regular soft stool). Attention to adherence with PERT is valuable. A useful discussion and management algorithm for DIOS is described by Groves and colleagues.15
Children with CF are just as susceptible to viral gastroenteritis as their healthy paediatric peers and will present with the same clinical signs. Regular salt replacement dosing (oral electrolyte powder in infant and toddlers and table salt or salt tablets in older children) should be increased for the duration of the illness. Alternatively, certain oral electrolyte hydration preparations can be used in lieu. However, when investigating children with isolated diarrhoea, differential diagnoses to consider include insufficient PERT dosing (steatorrhea rather than diarrhoea), constipation-related overflow, Clostridium difficile symptomatic infections and small intestinal bacterial overgrowth.
Cystic fibrosis liver disease
Cystic fibrosis liver disease (CFLD) is a very common complication of CF.16 CFTR is expressed in the hepatic ducts and regulates bile flow. Most adults have biochemical evidence of liver involvement, although clinically significant liver disease with portal hypertension and liver cirrhosis manifests in 5 to 10% of children. Almost all serious cases are detected within the first two decades of life.17After pulmonary and lung transplant complications, CFLD is the third highest cause of deaths in people with CF.
CFLD is characterised by a progression from hepatic steatosis (almost all patients) to focal biliary cirrhosis and, finally, multilobular cirrhosis. Impaired CFTR function leads to inspissated bile accumulation in hepatic ducts, with subsequent hepatocyte injury, and portal tract inflammation and fibrosis.17 Patients with advanced CFLD develop portal hypertension with splenomegaly and often hypersplenism. They rarely progress to severe synthetic failure. Severe CFLD may interfere with nutrition through inadequate secretion of bile salts. CFLD is an independent risk factor for the development of CF-related diabetes (discussed in the next part of this series).
Most units screen for CF-associated liver disease with annual blood tests (liver function tests) and biennial liver ultrasounds from about the age of 5 years. However, there is little proven, effective therapy for CFLD. Ursodeoxycholic acid may benefit asymptomatic patients with early-stage liver disease.18 Some patients with CF have had liver transplants.
Conclusion
GPs have a significant role to play in the nutritional management of patients with CF. Adherence with PERT and salt and vitamin replacement is vital and behavioural eating problems are a common feature to deal with. Recurrent abdominal pain is a common reason for patients with CF to present to the GP and there are many CF-specific and general causes to consider. Through it all, a close relationship with the GP can provide invaluable moral support amid the complexity of this chronic illness.
The final article in this series will examine endocrine issues including CF-related diabetes, sexual health, mental health and prevention medicine. RMT
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.