Paediatric cystic fibrosis. Part 2: Pulmonary management

Dr Pham is a Paediatric Respiratory Fellow and Professor Massie is a Paediatric Respiratory Consultant at the Royal Children’s Hospital, Melbourne, Vic.
Cystic fibrosis
Lung diseases
Lung disease is a common problem in people with cystic fibrosis. Daily treatment to control airway infection, delay development of bronchiectasis and limit pulmonary exacerbations primarily involves airway clearance, mucolytic therapy and antibiotics, but new medications are also helping a growing number of patients.
- Cystic fibrosis (CF) lung disease is a result of airway infection, inflammation and mucociliary obstruction, with structural airway changes occurring from an early age.
- Daily treatment with airway clearance and mucolytic therapy aims to delay the progression of bronchiectasis.
- Pulmonary exacerbations are often managed in an outpatient setting with antibiotics and additional airway clearance.
- Treatment with CF transmembrane conductance regulator modulators lowers sweat chloride levels, improves lung function and reduces pulmonary exacerbations for CF patients with responsive mutations.
- CF teams encourage the involvement of GPs in the care of patients with CF and are readily available to guide management decisions.
Pulmonary disease is a major cause of morbidity and mortality in people with cystic fibrosis (CF). Lung disease starts early in those with CF, with 60% of children having confirmed evidence of bacterial infection by 6 months of age and 40% having bronchiectasis confirmed on CT scans by 3 years of age.1
The aim of this article is to familiarise GPs with the pulmonary treatment of CF, particularly in children. It covers the pathophysiology of CF lung disease, daily treatment (including new medications that activate the mutated protein, the CF transmembrane conductance regulator [CFTR]) and management of pulmonary exacerbations. The previous article in this series covered diagnosis of CF and following articles will examine nutritional management and general care, such as vaccinations, smoking cessation (active and passive exposure) and sexual and mental health.2
Pathophysiology of lung deterioration
The lungs of people with CF are normal at birth, but there is a cycle of infection, inflammation and structural airway changes. This intricate relationship is an ongoing process that has been documented by the Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF).3,4 The goal of treatment is to slow the progression of airway disease and control respiratory symptoms.
Normal respiratory airways are lined by a thin airway surface liquid, which is regulated by transport of electrolytes (principally chloride and sodium) through the CFTR. An absent or minimal-function CFTR causes a desiccated airway surface liquid that traps bacteria and impairs mucociliary clearance. The small airways become obstructed, and chronic inflammation damages the airways, eventually causing bronchiectasis (Figure). Staphylococcus aureus is the most common organism implicated in CF lung disease, with more than half of individuals with CF having at least one positive culture for methicillin-sensitive S. aureus and 25% having multidrug-resistant S. aureus. Pseudomonas aeruginosa is also a leading cause of airway infection (45%), with 18% of strains being multidrug-resistant. Other bacteria found in people with CF include Haemophilus influenzae, Stenotrophomonas maltophilia, Achromobacter xylosoxidans and Burkholderia cepacia.5 Certain fungi and nontuberculous mycobacteria are becoming more common as a cause of CF pulmonary exacerbations, and pose challenging treatment dilemmas.6,7 CFTR dysfunction also alters the innate host defence and, of itself, contributes to pulmonary inflammation.
{kind=link}
Exacerbations of CF lung disease are common but poorly understood. There is no doubt that viral infections cause increased mucus production and temporarily impair mucociliary clearance, but they also predispose to bacterial growth.
Much of the damage to airways in people with CF happens inexorably, and often quietly, over many years. Clinical exacerbations add to the evolving airway damage. Young children may have recurrent wheeze as a clinical expression of small airway obstruction. When airway damage has progressed to generalised bronchiectasis, a regular moist cough appears, although some patients and their families are so used to the cough that they do not report it. Extensive disease will be associated with limitation of physical activity and often nutritional difficulties, as the metabolic load of chronic infection increases. Eventually, the progressive bronchiectasis leads to respiratory failure and is the main cause of death in people with CF.8
Daily treatment of CF lung disease
The goals of respiratory treatment in people with CF are to:
- delay development of bronchiectasis
- control chronic airway infection
- limit or shorten pulmonary exacerbations.
The foundations of daily treatment for CF lung disease are airway clearance, mucolytic therapy, antibiotics and, for an increasing number of patients, the new medications that activate CFTR. All these treatments help delay the development of bronchiectasis or slow its progression.
Airway clearance
Regular airway clearance is a fundamental part of daily CF care. For infants and young children, this is delivered as chest percussion. From the age of 3 years, most children can do some form of positive expiratory pressure (PEP) technique, such as blowing bubbles (‘bubble PEP’). Older children use various devices, including PEP masks (with or without an attached nebuliser device) and airway oscillating devices. There is limited evidence to support the use of one airway clearance technique over another, but patients who can do regular airway clearance generally have fewer exacerbations and better lung function.9
Patients are encouraged to perform airway clearance for 20 minutes once or twice daily, stepping up the frequency during pulmonary exacerbations. They are also strongly encouraged to complement airway clearance with regular aerobic exercise. Younger children often enjoy trampolining as an adjunct to airway clearance.
Mucolytic therapy
Dornase alfa is the mainstay of mucolytic therapy. It is a recombinant human monoclonal deoxyribonuclease that cleaves neutrophil nucleic acids, which form the sticky part of CF mucus. Dornase alfa improves lung function and reduces pulmonary exacerbations.10 Nebulised dornase is administered once daily and can be used from infancy if there is evidence of lung damage on imaging.
Nebulised hypertonic saline 3 to 6% or inhaled mannitol dry powder can be used to increase the osmotic gradient across the epithelial layer and improve mucociliary clearance. Their effect is not as good as that of dornase alfa in sustained use, but either can be a useful adjunct to treatment. Some patients will use hypertonic saline or mannitol dry powder regularly, whereas some will add it only during pulmonary exacerbations. For many patients with CF, adding further therapies comes with a time cost, which may limit adherence. Patients with CF who also have reactive airways generally preload with salbutamol.
Antibiotic treatment
Regular antibiotic use varies between CF centres. In general, patients with established chronic infection (bronchiectasis and positive results of sputum culture) will use daily oral or inhaled antibiotics (e.g. tobramycin), depending on airway microbiology. Some CF units use antibiotic prophylaxis during the first few years of life because of the concerns about early airway damage from Haemophilus and Staphylococcus species, although there are no randomised trial data to support this practice.11 It is unclear how to best use antibiotics for patients without symptoms who have positive microbiological test results. Antibiotic use is essential when patients develop a new cough or have an exacerbation.
Antibiotic selection will be guided by previous culture results. If no results are available, an antibiotic with coverage for H. influenzae and S. aureus (e.g. amoxycillin-clavulanic acid, cefuroxime or trimethoprim-sulfamethoxazole) is a good starting point while waiting for a sputum or cough swab result. When P. aeruginosa is first cultured (or after a long period of negative culture results), it is usually possible to eradicate it from the airway with two months of nebulised tobramycin treatment. Patients chronically infected with P. aeruginosa usually receive a regular program of antipseudomonal therapy (e.g. nebulised tobramycin or inhaled dry-powder tobramycin). Ciprofloxacin is the only oral antipseudomonal antibiotic available, but the risk of developing resistance is high, even after a couple of courses. As such, it is used intermittently or as part of a rotation of antipseudomonal therapy, for no more than a month at a time.
Other treatments
Although inflammation is an important factor in CF lung disease, there are limited anti-inflammatory therapies (e.g. ibuprofen or corticosteroids). Evidence is weak for the benefits of ibuprofen, and the benefits must be balanced against the risk of serious gastric bleeding. Similarly, the benefits of corticosteroids are likely outweighed by the risk of growth suppression and cataracts. Inhaled corticosteroids have no benefit.12
Patients chronically infected with P. aeruginosa have been shown to experience a small improvement in lung function with regular azithromycin given three times weekly.13 The mechanism of action of azithromycin is unclear, but it has been shown to reduce production of bacterial virulence factor and biofilm, and it has some bactericidal effects.
Medications that modulate CFTR
The advent of CFTR modulators has been an exciting advance in CF care. There are three main types of CFTR modulators: potentiators (ivacaftor), correctors (lumacaftor and tezacaftor) and amplifiers (Table). CFTR potentiators improve gating properties of some specific types of mutated CFTR, improving electrolyte transport. CFTR correctors allow better folding of mutated CFTR so it can pass through the epithelial cell cytosol without being destroyed by cellular quality control mechanisms. In clinical use, correctors are paired with a potentiator to improve mutated CFTR gating function at the epithelial surface. CFTR amplifiers are under development, with the proposed aim of increasing CFTR production from the epithelial cell nucleus.
{kind=link}
The available CFTR modulator medications are tablets or granules (for those <25 kg) that are taken twice daily. They have a high annual cost per patient (up to $270,000), but patients who qualify under the Section 100 Highly Specialised Drugs program of the PBS (based on their age and responsive CFTR gene mutations) receive them at a subsidised rate. Patients with CF have healthcare cards and pay $6.30 per script. Ivacaftor is used for patients over 2 years of age with gating mutations, the most common being the G551D mutation. The lumacaftor-ivacaftor combination is licensed for use in patients from 6 years of age who are homozygous for the p.F508del mutation. A combination of tezacaftor-ivacaftor has recently been approved and is indicated for the treatment of patients with CF aged 12 years and older who are homozygous for the F508del mutation or who have at least one mutation in the CFTR gene that is responsive to tezacaftor-ivacaftor based on in vitro data and/or clinical evidence.
Treatment with these medications has been shown to reduce sweat chloride levels and improve clinical outcomes, including improvements in lung function, fewer exacerbations, better nutrition and better quality of life. They have few side effects, with some patients experiencing a transient dyspnoea on medication initiation and abnormalities in liver function test results.14 These are practical examples of genuine personalised medicines, with therapy targeted at specific CFTR gene mutations.
Next-generation CFTR modulators include triple combinations that will benefit patients who are heterozygous for the p.F508del mutation (regardless of the second mutation). Once these are available, there will be CFTR modulator medication suitable for most people with CF.
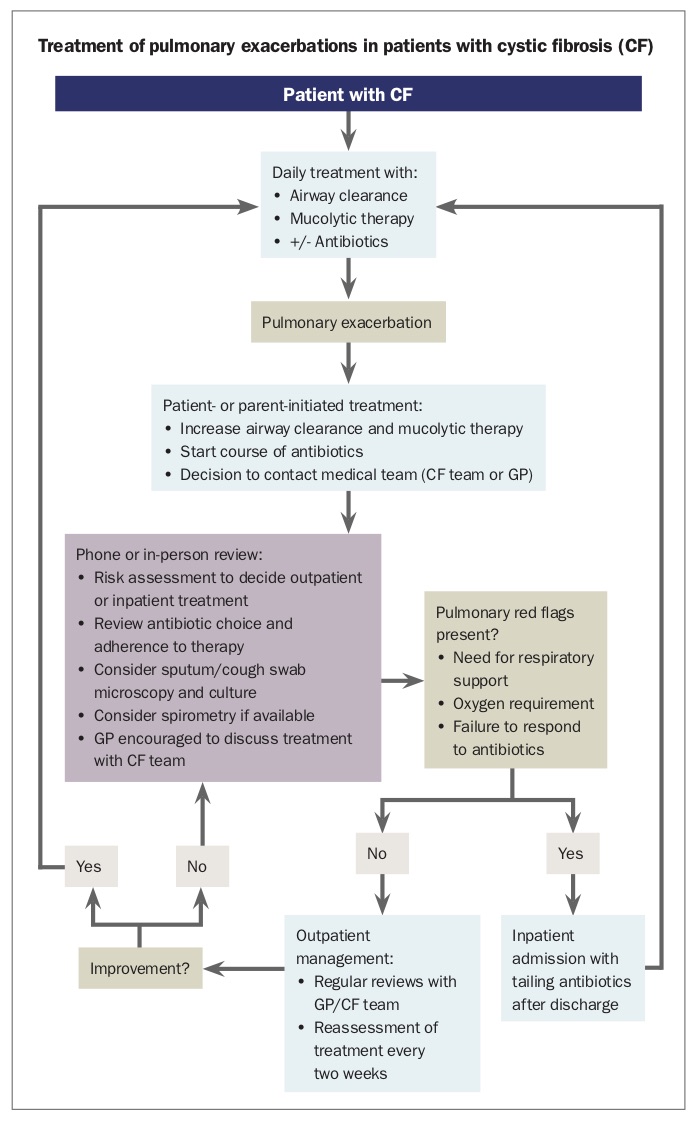
Management of pulmonary exacerbations
A CF pulmonary exacerbation is defined as a substantial change from baseline. This may be a change from no cough at baseline to a daily cough, or from a regular early morning cough with minimal sputum production to a more substantial cough in terms of sputum production, duration and time of occurrence during the day (and night). Reduced exercise capacity may be a sign of worsening lung function. Additional systemic symptoms of haemoptysis, fever, loss of appetite or weight loss may indicate more unwell patients. Whether the symptoms are caused by a gradual worsening of CF lung disease or in response to a viral illness may influence management.
On examination, work of breathing with use of accessory muscles, respiratory rate and chest auscultation may be normal during exacerbations. The patient should be asked to ‘huff’ and cough; a change in the quality of cough from baseline is a positive finding. If spirometry is available, a fall in the measurement of forced expiratory volume in one second of 10% or more from baseline is likely to represent a significant exacerbation. Peak expiratory flow is too variable to rely on as a monitoring tool. Collection of sputum (or a cough swab from those too young to produce sputum) after about one week of persistent symptoms is helpful to guide antibiotic management. Chest radiography may be warranted if there are new localised signs, remembering that CF is an airway disease rather than a recurrent airspace disease (i.e. pneumonia).
Most patients with exacerbations can be managed in an outpatient setting. Antibiotics are essential; if patients use antibiotics on an ‘as required’ basis, they are encouraged to start a course when their cough increases from its baseline level, particularly when there is a change in the characteristics of the cough or increased sputum productivity. The choice of antibiotic is often guided by previous microbiological growth. If patients are already taking regular antibiotics, collecting a sputum sample may guide a change. Antibiotics are essential even if the exacerbation is obviously viral, as the virus creates an airway milieu conducive to bacterial infection, and evidence is emerging of the way viruses may promote quiescent bacteria to grow. Patients should also increase the frequency of their airway clearance to twice daily and could add a mucolytic agent, such as hypertonic saline, if they are not already using one regularly.
The patient should be reassessed after a week or so to check clinical progress, perform or follow up sputum cultures and remeasure lung function. However, it is not uncommon for exacerbations to last for two to four weeks. Hospital admission may be warranted if a patient’s condition fails to improve despite multiple courses of oral antibiotics or if they develop increasing respiratory distress, haemoptysis or systemic symptoms. Each CF service will have a clinical nurse consultant who knows the patient well, or a physician on call, who can discuss patient progress (Flowchart). Families and GPs are encouraged to contact their CF team during pulmonary exacerbations.
{kind=link}
Hospital treatment includes intravenous antibiotics, physiotherapist-guided airway clearance sessions and respiratory support, including oxygen and noninvasive ventilation if indicated. Patients are often discharged home on a tailing course of antibiotics before restarting their usual regimen.
The role of the GP
The GP has an important role to play in the management of CF lung disease. GPs will know the patient’s baseline, have a strong therapeutic relationship with them, know their social circumstances and provide easy access to care. They are instrumental in facilitating adherence to the rigorous daily treatment regimen and in early detection of pulmonary exacerbations. GPs can also provide advice about smoking cessation and facilitate annual influenza vaccinations for patients and their families.
Conclusion
Pulmonary complications cause significant morbidity for patients with CF. Daily treatment with airway clearance, mucolytic therapy and aggressive antibiotic treatment aims to control the chronic cycle of pulmonary infection, inflammation and mucociliary obstruction to delay airway structural changes. The advent of CFTR modulators that directly target CFTR function holds promise for long-term treatment. Working alongside CF teams, GPs should feel empowered in the care of their patients with CF.
The following articles in this series will examine nutritional management, preventive management, sexual and reproductive health and mental health. RMT
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.