Eosinophilic lung diseases: an overview
Dr Han and Dr Di Michiel are Respiratory and Sleep Physicians in the Department of Respiratory Medicine, Concord Repatriation General Hospital, Sydney. Clinical Professor Morgan is a Respiratory Physician in the Department of Respiratory Medicine, Concord Repatriation General Hospital and Respiratory and Sleep Medicine, Nepean Hospital; and Clinical Professor in the School of Medicine, The University of Sydney, Sydney, NSW.
Lung diseases
Asthma
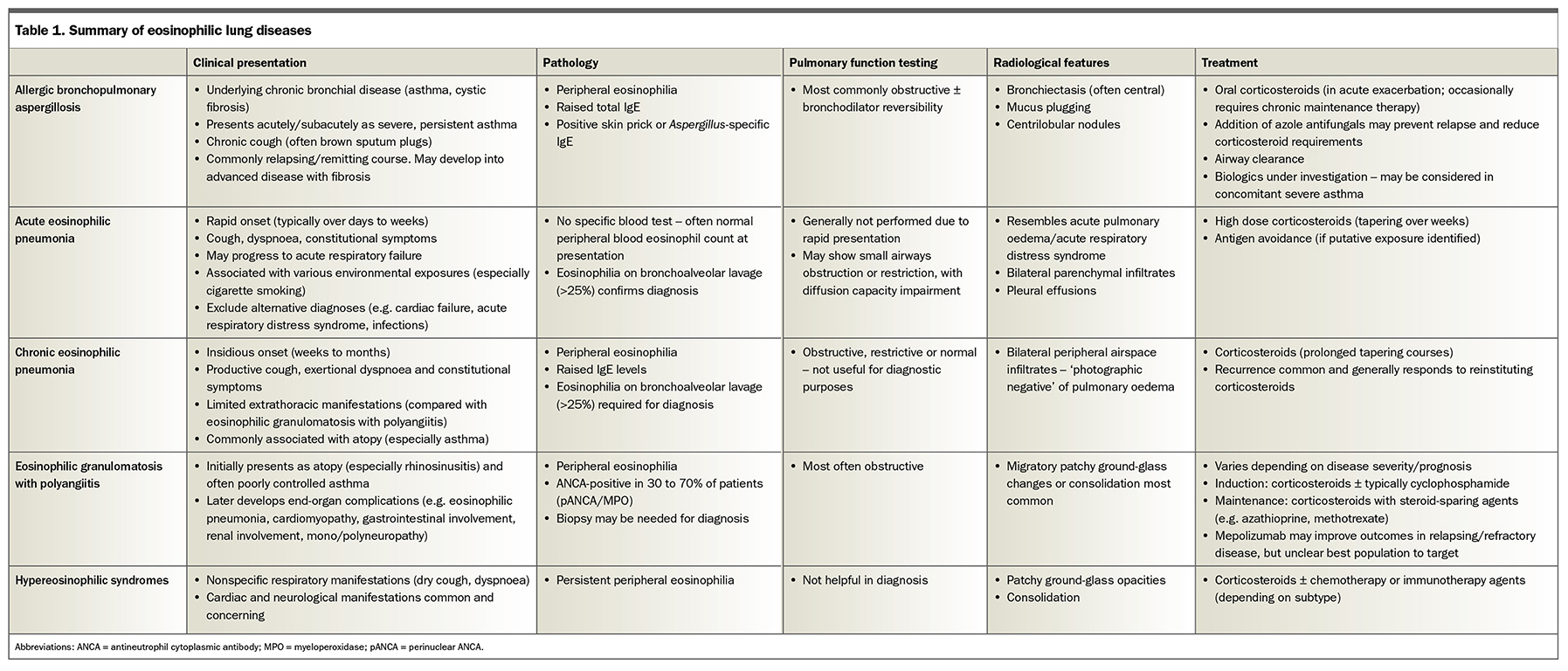
Eosinophilic lung diseases are characterised by elevated eosinophil levels with associated clinical, laboratory and radiographic features and include allergic bronchopulmonary aspergillosis, acute and chronic eosinophilic pneumonias, eosinophilic granulomatosis with polyangiitis and hypereosinophilic syndromes. The role of eosinophilic inflammation in other respiratory conditions (including asthma, chronic obstructive pulmonary disease and bronchiectasis) is increasingly recognised. Corticosteroid therapies are the cornerstone of treatment, and biologic agents now have an established role in the management of severe asthma and are under investigation in other eosinophilic lung diseases.
- Other causes of peripheral eosinophilia, such as allergic disease, drug reaction and parasitic infection, should be excluded before a diagnosis of eosinophilic lung disease is made.
- Specific clinical features may support diagnosis of particular conditions, for example, extrathoracic manifestations may suggest eosinophilic granulomatosis with polyangiitis, whereas recurrent severe asthma with evolving bronchiectasis may suggest allergic bronchopulmonary aspergillosis.
- Most forms of eosinophilic lung disease are highly responsive to corticosteroid therapy.
- The expanding role of biologic therapies targeting eosinophilic inflammation is an exciting area for future research in the management of these conditions.
Eosinophils are granulocytic white blood cells that are involved in immune responses to infection, tissue damage and tumour surveillance. Serum eosinophilia is seen in a variety of pathologies, including allergic diseases, drug reactions and infections (especially parasitic infections), but may also be a marker of underlying lung disease.1 Eosinophilic lung diseases are a heterogenous group of conditions characterised by elevated eosinophil levels (in either peripheral blood, bronchoalveolar lavage [BAL] specimens or lung tissue) associated with characteristic clinical, laboratory and radiographic abnormalities. This review discusses how to distinguish between and manage some of these conditions (summarised in Table 1), as well as outlining the role of eosinophilic inflammation in asthma, chronic obstructive pulmonary disease and bronchiectasis.
{kind=link}
Asthma
It is now well established that asthma is a heterogenous disease with multiple phenotypes and endotypes. Categorisation into inflammatory phenotypes such as allergic asthma, characterised by increased IgE levels, and eosinophilic asthma, with increased blood or sputum eosinophils, is useful in directing therapy. Eosinophilic asthma is associated with adult-onset disease and a more severe, often corticosteroid-dependent phenotype.2 Interleukin-4 (IL-4) and interleukin-5 (IL-5) are potent pro-eosinophilic cytokines that contributes to airway inflammation in asthma. In addition to inhaled corticosteroids (ICSs) and bronchodilators, monoclonal antibody therapies targeting the IL-4 and IL-5 pathways are now well established in the management of severe asthma. These ‘biologics’, including mepolizumab (anti-IL-5 monoclonal antibody), benralizumab (anti-IL-5 receptor monoclonal antibody) and dupilumab (anti-IL4 receptor alpha-subunit monoclonal antibody), have been shown to reduce asthma exacerbation rates and oral corticosteroid requirements among patients with severe eosinophilic asthma.3 Biologics are recommended in Australia for the management of patients with severe asthma despite optimal asthma therapy (including with high-dose ICS and oral corticosteroids), who have peripheral blood eosinophilia (defined as eosinophil count 0.30 × 109/L or ≥0.15 × 109/L in patients on oral corticosteroids).4 These medications are given as subcutaneous injections, usually every four to eight weeks, and are typically well tolerated.
Chronic obstructive pulmonary disease
Among patients with chronic obstructive pulmonary disease (COPD), those who exhibit peripheral blood eosinophilia experience more frequent exacerbations and a greater response to ICS therapy.5 International COPD management guidelines suggest using blood eosinophil count as a biomarker for the efficacy of add-on ICS therapy in long-term COPD management; patients with a blood eosinophil count of above 0.3 × 109/L have the greatest likelihood of treatment benefit.6
The role of biologic therapies in COPD is less clear than in asthma. A report of two randomised controlled trials of mepolizumab in patients with COPD and peripheral blood eosinophilia showed a reduced rate of exacerbation compared with placebo in one trial and no effect in the other.7 A post-hoc analysis showed a benefit for mepolizumab use compared with placebo on exacerbation rates among patients with a peripheral blood eosinophil count above 0.3 × 109/L.7 Benralizumab has not been shown to affect rates of COPD exacerbations compared with placebo.8 Although no biologic agents are currently approved for COPD management in Australia, this research gives a tantalising insight into a potential future of targeted therapies for patients with severe COPD.
Allergic bronchopulmonary aspergillosis
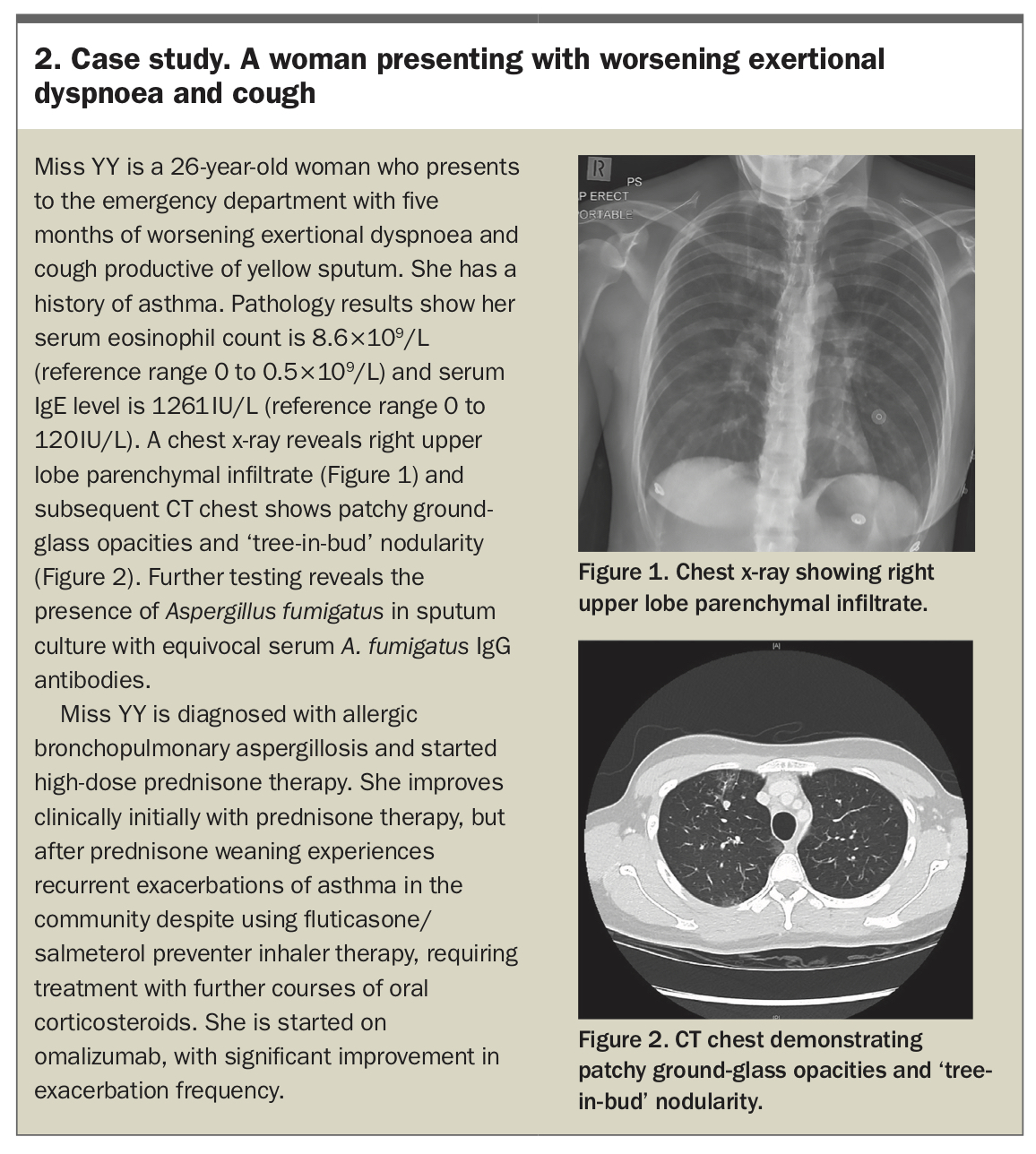
Allergic bronchopulmonary aspergillosis (ABPA) involves an intense eosinophilic hypersensitivity response to Aspergillus fungal antigens.9 ABPA typically occurs in patients with a history of chronic bronchial disease, especially asthma and cystic fibrosis. The natural history of ABPA is variable, and several staging criteria for clinical progression have been proposed.10,11 Initially, ABPA presents acutely or subacutely with a severe, persistent, often steroid-dependent asthma associated with chronic expectoration of sputum (sometimes containing plugs) – constitutional symptoms may be a presenting symptom in early disease. An initial clinical response to treatment is often complicated by disease relapse as treatment is weaned. Over time, some patients develop a chronic corticosteroid-dependent ABPA, whereas others progress to advanced ABPA with associated fibrosis. In the acute setting, peripheral eosinophilia (>0.5 ×109/L) and markedly elevated total serum IgE levels (typically >1000 kU/L) should raise the suspicion of ABPA in patients with supportive clinical features; in such patients, skin prick testing or detection of serum IgE/IgG antibodies against Aspergillus fumigatus support the diagnosis.11 Radiographic features on high-resolution chest CT include central bronchiectasis (with upper and middle lobe predilection), centrilobular nodules and mucous plugging.11 Diagnosis is made based on International Society for Human and Animal Mycology (ISHAM) criteria (Box 1).11
{kind=link}
The cornerstone of treatment is systemic corticosteroids during the acute phase, followed by slowly tapering of the course depending on clinical response.12 Serum IgE levels provide a useful indicator of treatment response. Recurrence after corticosteroid discontinuation is frequent. Consideration of the addition of oral azole antifungal agents (such as itraconazole or voriconazole) to corticosteroid therapy, either in the acute phase or for patients who relapse on corticosteroid monotherapy, is recommended to prevent recurrence and reduce corticosteroid requirements.13 Azole antifungal monotherapy has shown effectiveness against ABPA in smaller randomised controlled trials, but needs verification in larger trials.14,15
Biologics, including omalizumab and mepolizumab, have shown efficacy in treating ABPA, with one small randomised trial of omalizumab showing reduced exacerbation frequency compared with placebo.16-18 Currently, biologics may be considered for treatment of ABPA in patients with concomitant asthma, after respiratory specialist review. Nonpharmacological management, including airway clearance, pulmonary rehabilitation, regular vaccination and treatment of comorbid conditions, is essential.13 A case study outlining the management of a patient presenting with ABPA is presented in Box 2.
{kind=link}
Bronchiectasis
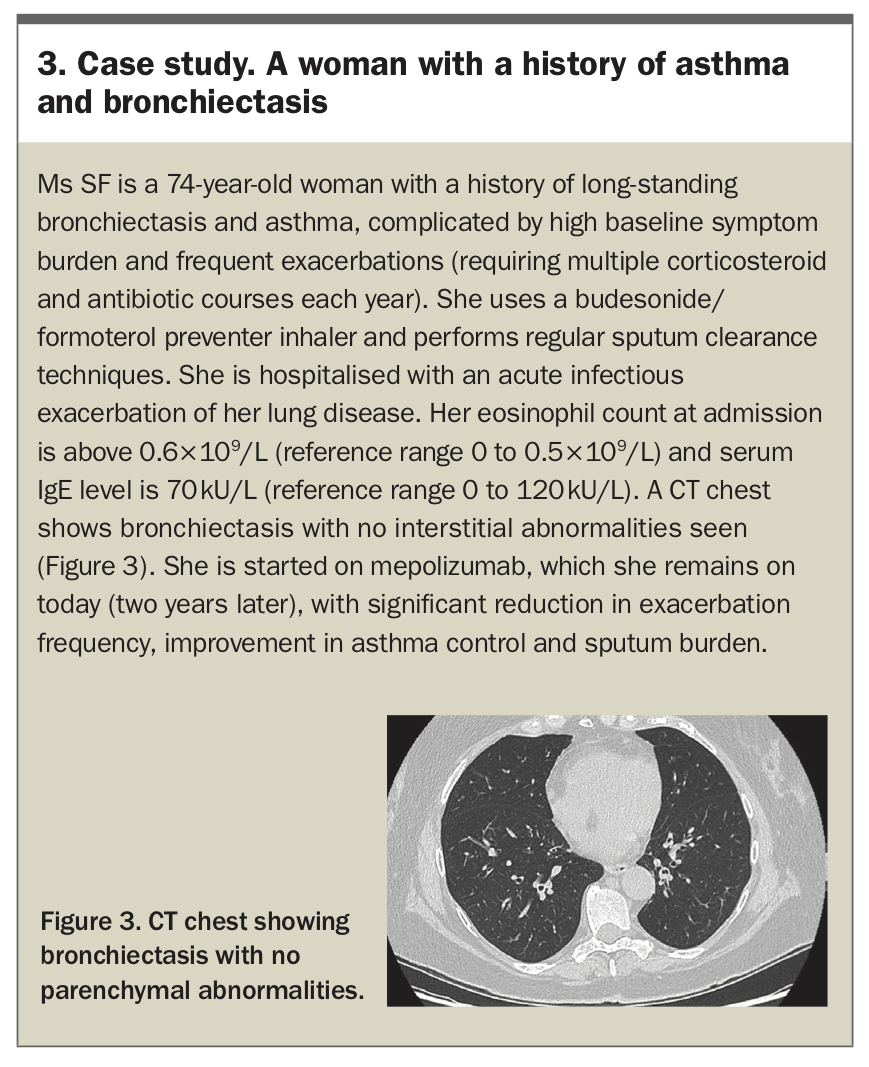
In addition to the effect of eosinophilic inflammation on the development of bronchiectasis in ABPA, the overall role of eosinophils in patients with bronchiectasis is an area of increasing research. In non-cystic fibrosis bronchiectasis, associations between serum and sputum eosinophilia, bronchodilator reversibility on spirometry and risk of exacerbations have been reported.19-20 Currently, ICS therapy in patients with bronchiectasis is only recommended for those with coexisting obstructive airways disease, with recent studies on the role of peripheral eosinophilia as a predictor of ICS response in patients with bronchiectasis showing mixed results.21-23 Eosinophil-targeting biologic agents (including mepolizumab and benralizumab) have been reported to improve spirometry, symptoms and quality of life among patients with poorly-controlled bronchiectasis and peripheral blood eosinophilia; however, this finding requires replication in larger studies.24 A case study of a patient with asthma and bronchiectasis is presented in Box 3.
{kind=link}
Acute eosinophilic pneumonia
Acute eosinophilic pneumonia (AEP) is a potentially life-threatening form of pneumonia characterised by the rapid accumulation of eosinophils in the lung interstitium and alveoli. Patients typically present with cough, dyspnoea and constitutional symptoms developing over days to weeks. Progression is variable, ranging from paucisymptomatic disease that resolves spontaneously to life-threatening hypoxaemic respiratory failure.25 Although patients often have no history of allergic disease, multiple precipitants have been recognised, including cigarette smoke (the most frequently implicated trigger), inhalational agents (including drugs such as cocaine and marijuana), medications (especially antimicrobials, nonsteroidal anti-inflammatory agents and selective serotonin-reuptake inhibitors) and infectious agents (especially parasites and fungi).25
Radiographic findings of AEP are often indistinguishable from acute pulmonary oedema, with bilateral parenchymal infiltrates and pleural effusions often seen.26 Diagnosis requires ruling out other pathologies, such as cardiac failure, acute respiratory distress syndrome (ARDS) and infectious pathologies. Eosinophilia in BAL fluid is ideally needed to confirm the diagnosis, although the patient’s clinical status may limit the ability to obtain BAL fluid. No specific serum abnormalities are seen in patients with AEP, and peripheral blood eosinophil counts are often normal at presentation.
The cornerstone of treatment is high-dose corticosteroids, which typically produce rapid clinical and radiographic improvement, and are typically tapered over weeks. Recurrence is rare after successful treatment with corticosteroids.27 Antigen avoidance for patients in whom a putative exposure is identified is essential.25 A case study of a patient with AEP is presented in Box 4.
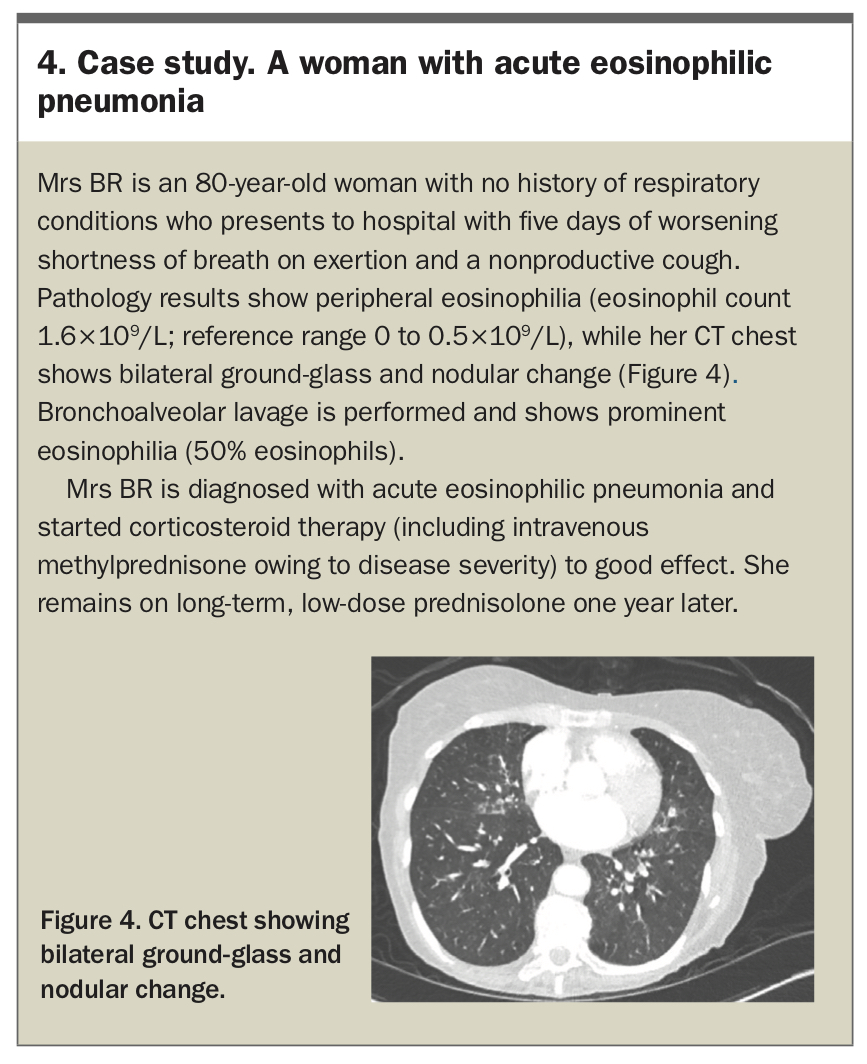{kind=link}
Chronic eosinophilic pneumonia
Chronic eosinophilic pneumonia (CEP) is an uncommon form of interstitial lung disease that typically presents with insidious onset of productive cough, dyspnoea and fevers, evolving over weeks to months. There is a strong association with atopy; in particular, asthma is reported in 50% of patients and allergic rhinitis is reported in 15 to 35%.28 Patients generally exhibit limited extrathoracic manifestations, in contrast with eosinophilic polyangiitis with granulomatosis (EGPA), an important differential. No laboratory tests are specific for CEP, but common findings include peripheral blood eosinophilia, raised inflammatory markers and elevated total IgE levels. Pulmonary function tests may show obstruction or restriction or be normal, and are not useful in clarifying the diagnosis. The classic radiographic appearance of CEP is a ‘photographic negative’ of pulmonary oedema with bilateral, peripheral and upper lobe-predominant airspace consolidation.26 However, this appearance is neither sensitive nor specific for CEP. In patients with convincing clinical and radiographic features of CEP, eosinophilia on BAL (>25%) confirms the diagnosis.29
CEP is highly responsive to treatment with systemic corticosteroids, with clinical and radiological improvement occurring within days. Disease recurrence is common and responds well to resumption of corticosteroids. The dose and duration are tailored to disease severity and corticosteroid side-effects – generally, six to 12 months of therapy is needed. Treatment of chronic eosinophilic pneumonia with biologic agents has been reported, but requires investigation in randomised controlled trials.30,31 Long-term prognosis for patients with CEP is excellent, with mortality rare and morbidity largely related to corticosteroid side-effects and persistent airway obstruction.29 A case study of a patient with CEP is presented in Box 5.
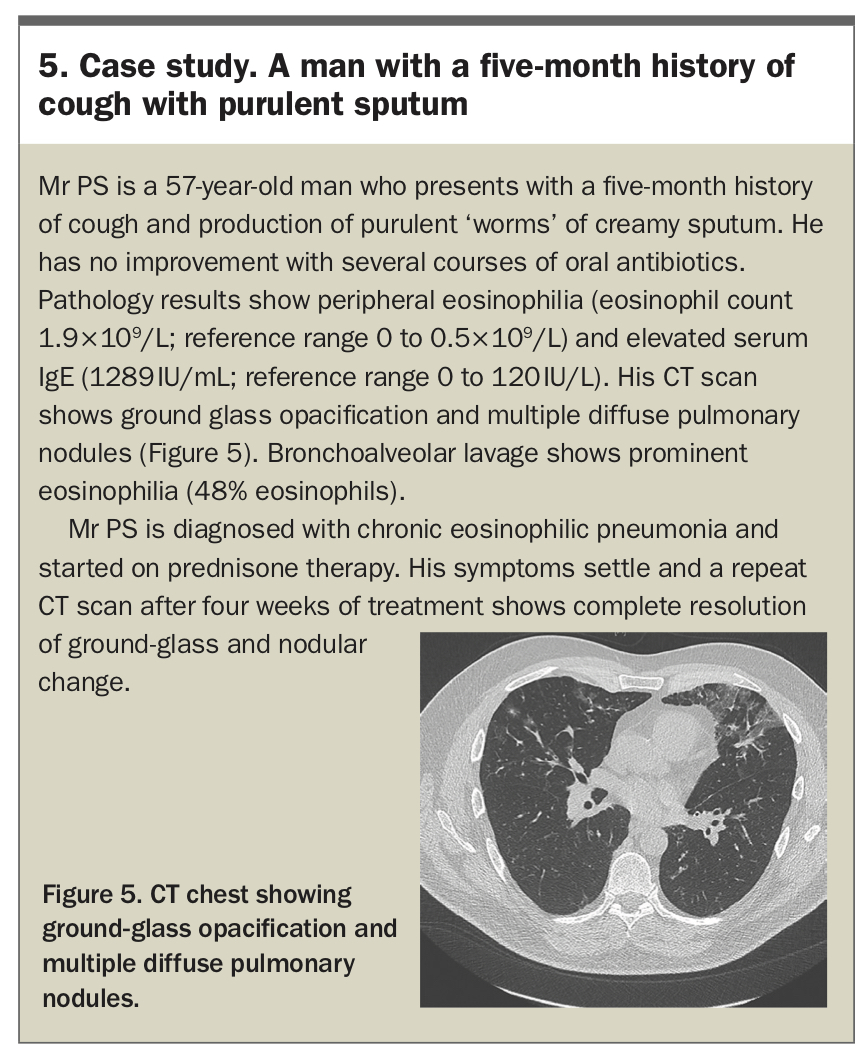{kind=link}
Eosinophilic granulomatosis with polyangiitis
Eosinophilic granulomatosis with polyangiitis (EGPA), previously known as Churg-Strauss syndrome, is a multisystem vasculitis characterised by asthma, rhinosinusitis and peripheral eosinophilia. Classically, EGPA is described as occurring in three phases. Initially, patients experience a prodromal phase, characterised by rhinosinusitis and an often-severe corticosteroid-dependent asthma. This is followed by an eosinophilic phase involving the development of peripheral blood and tissue eosinophilia, with associated end-organ effects, especially involving the lungs (eosinophilic pneumonia) and gastrointestinal tract. Finally, a vasculitic phase occurs, where patients develop features of systemic vasculitis involving skin, cardiac and neurological manifestations.32 However, there is often significant variability in clinical manifestations – in clinical practice, these phases may overlap, some patients may not develop eosinophilic or vasculitic manifestations at all, while in other patients any organ systems may be affected during either the vasculitic or eosinophilic phase of disease.33
Antineutrophil cytoplasmic antibody (ANCA) status affects disease manifestations. Patients who are ANCA-positive exhibit more frequent renal involvement, pulmonary haemorrhage and neuropathy, whereas patients who are ANCA-negative experience more frequent cardiac involvement.34 The end-organs involved (especially cardiac, gastrointestinal and renal involvement) are an important predictor of mortality and guide treatment decisions.33,34
Pathology shows peripheral blood eosinophilia (unless suppressed by corticosteroid therapy) and supports the diagnosis. Positive ANCA helps to confirm the diagnosis, and is typically of perinuclear ANCA (pANCA) class with myeloperoxidase (MPO) antibodies. However, ANCA is positive in only 30 to 70% of patients, and confirmatory biopsy showing a necrotising vasculitis may be required.35 Radiographic findings are often nonspecific, with patchy ground-glass changes or airspace consolidation. Progress imaging may show migratory infiltrates. Although several diagnostic criteria sets are available, the most up-to-date are the 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology (ACR/EULAR) criteria, summarised in Table 2.36
{kind=link}
Management of patients with EGPA depends on disease severity, which is typically determined using the Five Factor Score (age >65 years, cardiac involvement, renal insufficiency, gastrointestinal involvement and absence of ENT manifestations).37 Patients may receive induction therapy with either corticosteroid monotherapy in mild disease or combination intravenous cyclophosphamide/corticosteroid in more severe disease. Maintenance with corticosteroids and steroid-sparing agents, such as azathioprine and methotrexate, is generally needed – the optimal duration of maintenance therapy is unclear.35 Mepolizumab has been shown to improve rates and duration of remission in patients with relapsing or refractory EGPA and is indicated for this use in Australia; however, the best patient populations to target for therapy remain unclear.38 A case study of a patient with EGPA is presented in Box 6.
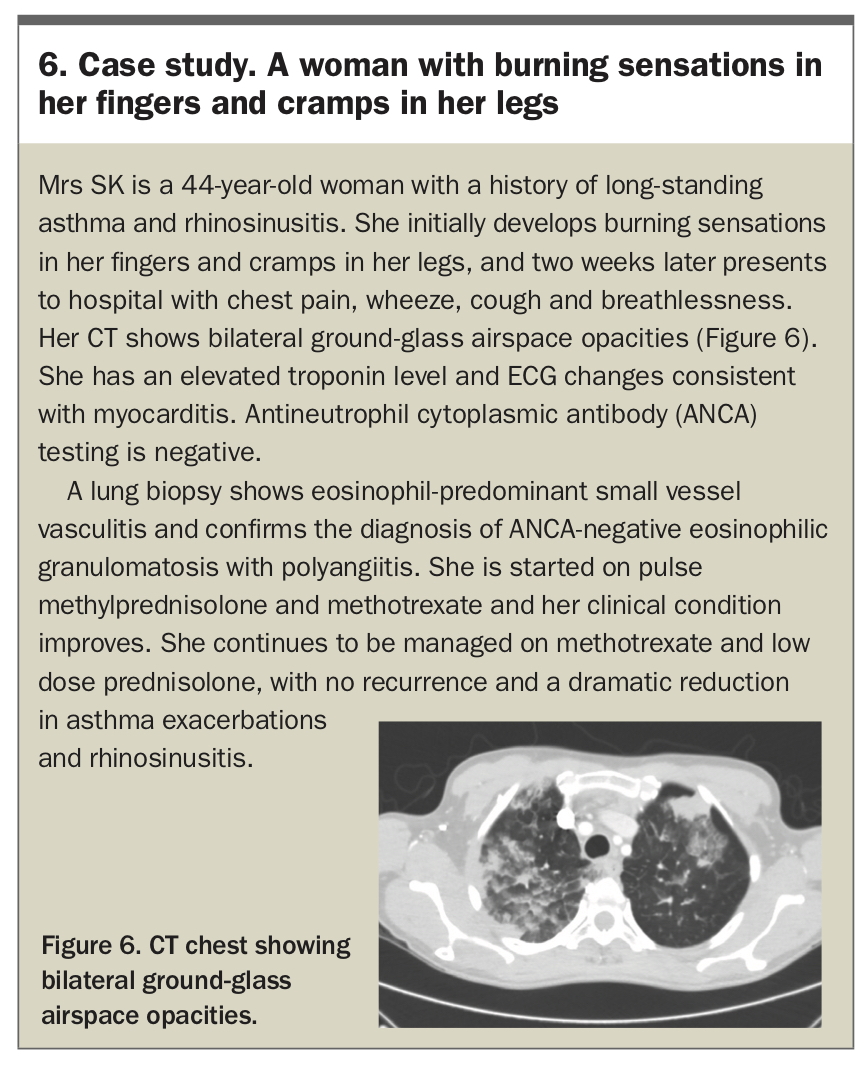{kind=link}
Hypereosinophilic syndromes
Hypereosinophilic syndromes (HESs) are characterised by a persistent peripheral eosinophilia (>1.5 × 109/L) without an identifiable cause, with associated features of eosinophilic end-organ involvement. Around half of cases are idiopathic, while specific subtypes including lymphocytic and myeloproliferative variants have been described and bone marrow aspiration is often required. Patients often develop cardiac and neurological manifestations, which are associated with significant morbidity and mortality. In contrast, respiratory manifestations are present in around 25% of patients and are often mild, with a nonspecific dry cough and possible shortness of breath.39 Radiographically, patients most often exhibit patchy ground-glass opacities and consolidation.40
Traditionally, treatment for idiopathic HES involves corticosteroid therapy, with treatment response varying by disease subtype.41 In recent years, there has been increasing interest in the use of monoclonal agents for treatment. Treatment of HES subtypes is complex and may involve chemotherapy and immunotherapy agents. A case study of a patient with HES is presented in Box 7.
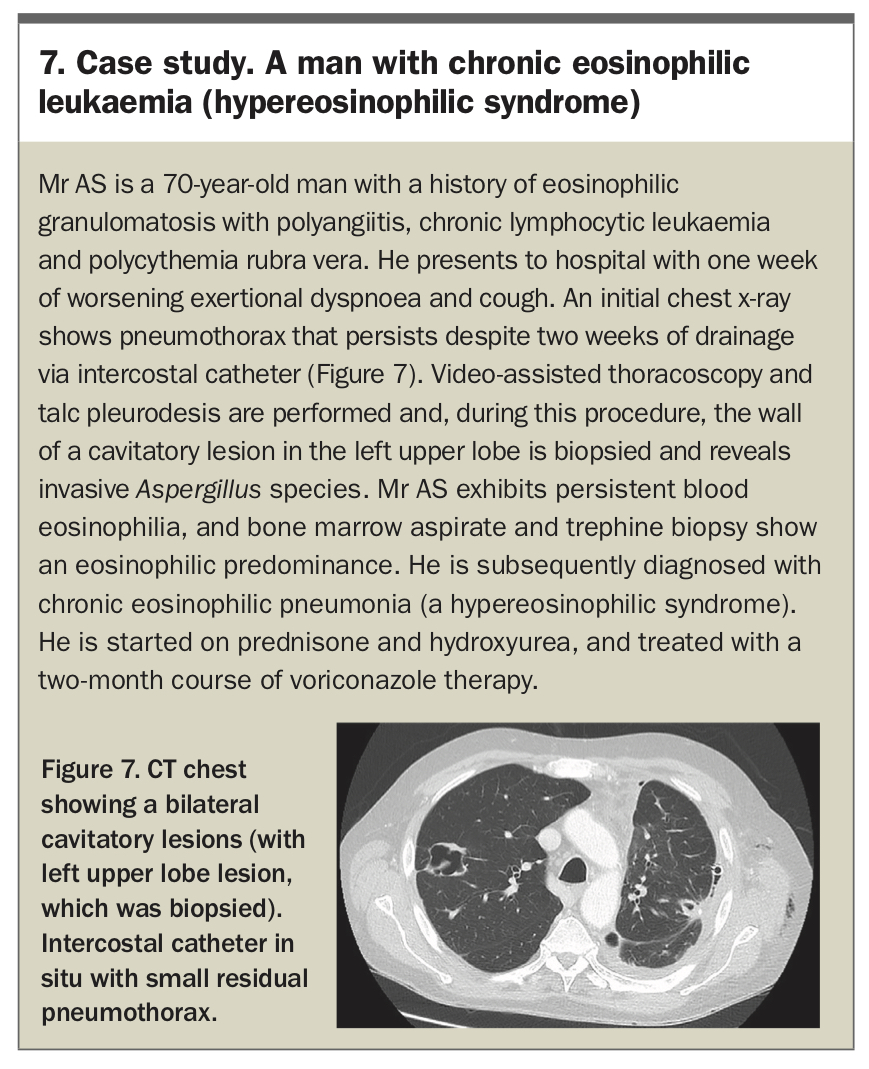{kind=link}
Conclusion
Eosinophilic lung diseases comprise of a heterogeneous group of conditions characterised by the presence of peripheral or tissue eosinophilia with pulmonary involvement. Although corticosteroid therapy is the cornerstone of treatment for many of these conditions, an understanding of the varied nature of eosinophilic lung disease presentations is essential to successful diagnosis and long-term management. RMT
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.